Cronkhite-Canada Syndrome: Causes, Symptoms, Diagnosis & Treatment
Comprehensive guide to Cronkhite-Canada syndrome: understanding rare gastrointestinal polyposis with dermatological manifestations.

Cronkhite-Canada Syndrome: Overview and Definition
Cronkhite-Canada syndrome (CCS) is a rare, sporadic disorder characterized by generalized gastrointestinal polyposis combined with distinctive dermatological manifestations. First described in 1955, this non-hereditary condition represents a significant clinical challenge due to its complex presentation and previously poor prognosis. CCS is defined by the association of non-neoplastic gastrointestinal hamartomatous polyps with a characteristic cutaneous triad comprising alopecia (hair loss), nail dystrophy, and hyperpigmentation.
Unlike hereditary polyposis syndromes, Cronkhite-Canada syndrome develops spontaneously and is not inherited. The condition affects individuals across all ethnic groups and typically manifests during adulthood, with symptom onset occurring at a mean age of 59 years. Since its initial description, more than 500 cases have been documented worldwide, making it an important consideration in the differential diagnosis of patients presenting with combined gastrointestinal and skin manifestations.
Demographics and Epidemiology
Cronkhite-Canada syndrome is a rare condition with sporadic occurrence. The typical age of symptomatic disease onset is approximately 59 years, although the syndrome can develop in individuals of various ages. The condition affects both males and females and has been documented across diverse ethnic populations, indicating no specific geographic or racial predilection.
The rarity of CCS means that awareness among healthcare providers remains limited, often resulting in delayed diagnosis. The non-hereditary nature of the syndrome means that family members of affected individuals do not carry increased risk of developing the condition, distinguishing it from familial polyposis syndromes.
Causes and Pathophysiology
The underlying cause of Cronkhite-Canada syndrome remains elusive despite decades of clinical observation and research. However, compelling evidence suggests an immune-mediated mechanism plays a central role in the disease pathogenesis. Supporting this hypothesis are several key findings: increased systemic levels of immunoglobulin G4 (IgG4) detected in CCS patients, elevated antinuclear antibody titers, and increased IgG4 mononuclear cell staining within CCS polyps themselves.
These immunological abnormalities suggest that CCS may represent an autoimmune condition affecting the gastrointestinal tract. The association of CCS with various autoimmune disorders further supports this mechanism. The polyps themselves demonstrate characteristic histological features with striking stromal edema and eosinophilic inflammation, distinct from typical juvenile-type polyps and supporting an inflammatory etiology.
The cutaneous manifestations of CCS, including the dermatological triad, appear to be secondary to malabsorption and malnutrition caused by the underlying gastrointestinal pathology. Dysgeusia (altered taste sensation) and xerostomia (dry mouth) may result from mucositis or oral infections secondary to nutritional deficiencies, though additional mechanisms may be involved in their pathogenesis.
Clinical Features and Presentation
Cronkhite-Canada syndrome presents with a combination of gastrointestinal and dermatological symptoms that typically develop in a characteristic sequence, though individual presentations may vary.
Gastrointestinal Manifestations
Diarrhea and taste disturbances are typically the earliest symptoms, often preceding dermatological manifestations. Diarrhea is usually profuse and persistent, reflecting the extensive gastrointestinal pathology. Dysgeusia (altered taste sensation) and hypogeusia (diminished taste) frequently occur as initial symptoms alongside or preceding other manifestations.
Patients commonly experience abdominal pain and discomfort, though the severity varies considerably. Weight loss is a frequent and often significant finding, resulting from diarrhea, malabsorption, and reduced nutritional intake due to altered taste sensation. The gastrointestinal symptoms may be accompanied by other complaints including xerostomia (dry mouth) and abnormal oral sensations.
Dermatological Manifestations
The characteristic cutaneous triad develops following the gastrointestinal symptoms:
- Nail dystrophy (Onychodystrophy): Nail changes develop as a variable sequence of manifestations including onycholysis (separation of nail from nail bed), thinning of the nail plate, onychoschizia (horizontal splitting of nails), and onychomadesis (shedding of nails). These changes typically affect both fingernails and toenails.
- Alopecia: Hair loss initially appears as patchy alopecia but progresses rapidly to complete baldness affecting the scalp. The progressive nature and extent of hair loss distinguish CCS-related alopecia from other causes.
- Hyperpigmentation: Cutaneous pigmentation changes manifest as diffuse light-to-dark brownish macules and plaques distributed on characteristic locations including palms and soles, upper extremities, face, and chest. Patchy vitiligo (depigmented areas) may also occur alongside hyperpigmentation. Pigmented macules may also appear on oral mucosa.
Most CCS patients exhibit all cardinal manifestations of the syndrome, though some individuals may present with incomplete features. The timing and sequence of symptom development can vary, with Japanese scholars classifying CCS into five clinical types based on presenting symptoms: Type I (diarrhea as initial symptom), Type II (hypogeusia as initial symptom), Type III (thirst or abnormal oral sensation), Type IV (abdominal discomfort other than diarrhea), and Type V (alopecia as initial symptom).
Laboratory Findings
Laboratory investigations typically reveal multiple abnormalities reflecting the systemic nature of the disease:
- Anemia (often iron deficiency or nutritional in etiology)
- Hypoproteinemia (low serum protein levels)
- Electrolyte disturbances
- Nutritional deficiencies (vitamins, minerals)
- Positive antinuclear antibody (ANA)
- Positive fecal occult blood test
- Elevated immunoglobulin G4 levels
Diagnosis and Investigations
Diagnosis of Cronkhite-Canada syndrome requires clinicopathologic correlation involving endoscopic, histopathological, and cutaneous features. The diagnosis should be considered in patients presenting with the combination of gastrointestinal hamartomatous polyps, diarrhea, and the characteristic dermatological triad.
Endoscopic Findings
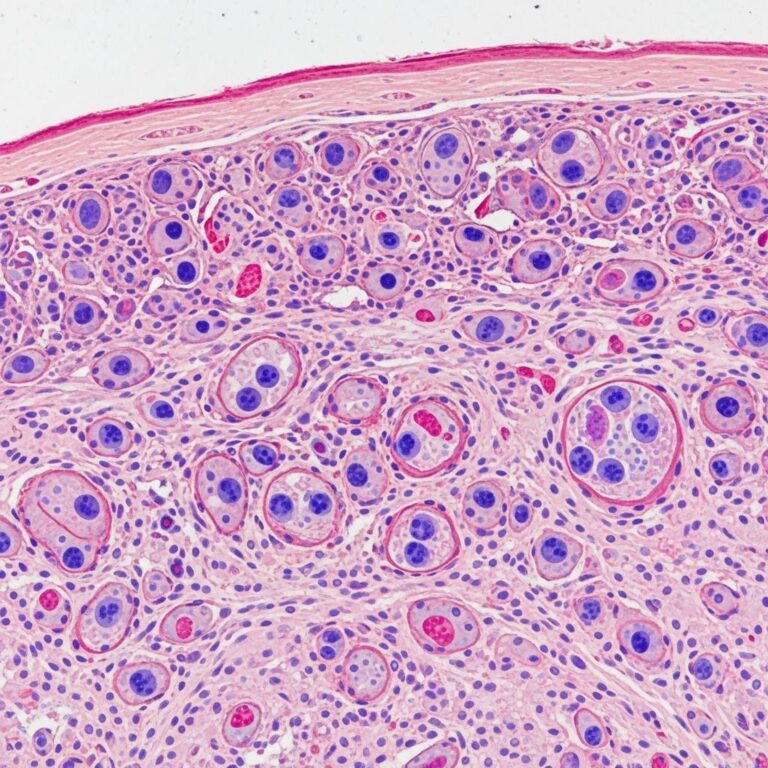
Upper endoscopy, colonoscopy, and small bowel imaging typically reveal diffuse polyposis throughout the gastrointestinal tract. Polyps are present in the stomach, small intestine, and colon but characteristically absent from the esophagus. The polyps are non-neoplastic hamartomas, distinguishing them from other polyposis syndromes. The widespread nature of polyps throughout the gastrointestinal tract is a key diagnostic feature.
Histopathological Features
Biopsy specimens demonstrate hamartomatous polyps with histology similar to juvenile-type polyps but with distinctive features. The stroma characteristically shows striking edema and eosinophilic inflammation, which is unique to CCS and helps differentiate it from other hamartomatous polyposis syndromes.
Additional Diagnostic Tests
Laboratory evaluation should include complete blood count, serum protein levels, electrolytes, nutritional markers, and serological testing including antinuclear antibodies and immunoglobulin levels. Fecal occult blood testing is typically positive. Carcinoembryonic antigen (CEA) levels should be assessed as part of cancer screening.
Differential Diagnoses
Several conditions must be distinguished from Cronkhite-Canada syndrome when patients present with gastrointestinal polyposis and/or dermatological manifestations:
- Juvenile Polyposis Syndrome: Develops before 10 years of age (earlier than CCS typical onset), characterized by hamartomatous polyps primarily in the colon with less involvement of stomach and small intestine, and lacks the characteristic dermatological triad of CCS.
- Hereditary polyposis syndromes: Distinguished by their hereditary pattern of inheritance, which is absent in CCS.
- Inflammatory bowel disease: Presents with diarrhea and gastrointestinal symptoms but typically without widespread hamartomatous polyps or the characteristic dermatological triad.
- Other dermatological conditions: Conditions causing alopecia, hyperpigmentation, or nail dystrophy must be differentiated by the presence of gastrointestinal polyposis and the specific clinical context.
Complications
Several serious complications can develop in patients with Cronkhite-Canada syndrome. Malignant transformation of CCS polyps may occur, with increased risk of colorectal cancer warranting aggressive screening in affected patients. Severe malnutrition and malabsorption can lead to significant weight loss and deficiency states affecting multiple organ systems. The disease is associated with high morbidity, and historically demonstrated high mortality, though this has improved substantially with modern treatment approaches.
Treatment and Management
Early recognition and aggressive treatment are crucial for improving outcomes in Cronkhite-Canada syndrome. The mainstay of medical treatment involves systemic corticosteroids, which have dramatically improved the prognosis of this previously devastating condition.
Corticosteroid Therapy
Standard corticosteroid regimen: The typical treatment protocol involves initiating prednisone at 40 mg daily for one week, followed by a 5 mg weekly reduction until discontinuation. In one study, symptomatic response was observed within three months in 10 of 11 CCS patients treated with this regimen, demonstrating excellent efficacy.
Early aggressive treatment with systemic steroids has dramatically improved survival rates and results in reversal of symptoms and signs within 2-4 months. Steroid dose reduction should proceed slowly, as rebound flare is common if dose reduction occurs too rapidly. Careful monitoring during the tapering phase is essential.
Steroid-Sparing Strategies
Relapse of symptoms is common during corticosteroid tapering, necessitating alternative approaches. Immunomodulatory agents, particularly azathioprine, have been employed as steroid-sparing therapy. In one study, five CCS patients responding to corticosteroid treatment were transitioned to azathioprine therapy (2 mg/kg/day), achieving maintenance of clinical remission with no relapse after approximately five years of follow-up.
Supportive Care
Nutritional support is essential, particularly in patients with severe malabsorption and weight loss. Total peripheral nutrition (TPN) has been utilized in patients with significant nutritional deficiency, resulting in weight gain and symptom improvement. Continuation of TPN depends on tolerance and clinical response.
Alternative and Adjunctive Therapies
Various other therapeutic agents have been attempted in CCS management, including H2-receptor antagonists, antibiotics, acid suppression therapy, cromolyn sodium, anabolic steroids, and surgical intervention, though corticosteroids remain the primary and most effective treatment. Combinations of these therapies may be employed based on individual patient response and tolerance.
Prognosis and Outlook
The prognosis of Cronkhite-Canada syndrome has undergone dramatic transformation with modern treatment approaches. Historically, CCS carried a poor prognosis with mortality rates approximately 50% in the first five years after diagnosis. Early aggressive treatment with systemic corticosteroids has substantially improved these outcomes.
With appropriate and timely treatment, most patients experience significant improvement or complete reversal of symptoms within 2-4 months. The transition to steroid-sparing immunomodulatory therapy has further improved long-term outcomes and quality of life. However, close follow-up and monitoring remain essential, as relapse can occur and regular endoscopic surveillance is recommended given the malignancy risk associated with gastrointestinal polyps.
Frequently Asked Questions
Q: Is Cronkhite-Canada syndrome hereditary?
A: No, Cronkhite-Canada syndrome is not hereditary. It is a sporadic, acquired condition that develops spontaneously and is not inherited from parents to children. Family members of affected individuals do not have increased risk of developing the condition.
Q: At what age does Cronkhite-Canada syndrome typically develop?
A: Symptom onset typically occurs at a mean age of approximately 59 years, though the condition can develop across a range of adult ages. It is not typical in children, though rare cases in younger individuals have been documented.
Q: How is Cronkhite-Canada syndrome diagnosed?
A: Diagnosis requires clinicopathologic correlation involving endoscopic findings of diffuse polyps throughout the gastrointestinal tract (except esophagus), histopathological confirmation of hamartomatous polyps with characteristic stromal edema and eosinophilic inflammation, and presence of the dermatological triad of alopecia, nail dystrophy, and hyperpigmentation.
Q: What is the primary treatment for Cronkhite-Canada syndrome?
A: Systemic corticosteroids are the mainstay of treatment. The standard regimen involves prednisone 40 mg daily for one week, followed by 5 mg weekly reductions. Most patients show symptomatic response within three months, with reversal of symptoms and signs occurring within 2-4 months.
Q: Can Cronkhite-Canada syndrome be cured?
A: With appropriate corticosteroid treatment and immunomodulatory therapy when needed, symptoms can be reversed and remission achieved. While curative outcomes have been reported, close monitoring is necessary as relapse can occur during treatment tapering or after discontinuation.
Q: What is the mortality rate for Cronkhite-Canada syndrome?
A: Historically, CCS had a 50% mortality rate in the first five years after diagnosis. However, early aggressive corticosteroid treatment has substantially improved survival rates, with most patients achieving good outcomes when diagnosed and treated promptly.
References
- Cronkhite-Canada syndrome — Orphanet (INSERM). 2024. https://www.orpha.net/en/disease/detail/2930
- Cronkhite-Canada Syndrome: An Acquired Condition of Gastrointestinal Polyposis and Dermatologic Abnormalities — Seshadri, P. et al. Gastroenterology & Hepatology. 2012-03. https://www.gastroenterologyandhepatology.net/archives/march-2012/
- Cronkhite–Canada syndrome: from clinical features to treatment — Gastroenterology Report, Oxford University Press. 2020-08. https://academic.oup.com/gastro/article/8/5/333/5917768
- Cronkhite-Canada Syndrome: A Case Report and Review of Literature — National Center for Biotechnology Information (NIH). https://pmc.ncbi.nlm.nih.gov/articles/PMC2732131/
Similar Articles






Read full bio of medha deb