Cutaneous Plasmacytosis Pathology: Key Insights Into Diagnosis
Comprehensive pathology guide to cutaneous plasmacytosis: clinical features, histopathology, diagnosis, and management insights.

Author: Patrick Emanuel, Dermatopathologist, Auckland, New Zealand.
Introduction
Cutaneous plasmacytosis is a rare dermatological condition characterized by the benign proliferation of mature plasma cells in the skin, often presenting with multiple red-to-dark-brown macules, papules, and plaques primarily on the trunk, face, neck, and axillae. This disorder, also referred to as cutaneous and systemic plasmacytosis (CSP) in cases with extracutaneous involvement, features polyclonal hypergammaglobulinemia and superficial lymphadenopathy without an identifiable underlying malignancy or infection. First described in Japanese populations, it has been increasingly reported worldwide, though remains exceedingly rare, particularly in non-Asian individuals.
The aetiopathogenesis remains poorly understood but may involve dysregulated cytokine production, particularly interleukin-6 (IL-6), which promotes B-cell proliferation, plasma cell differentiation, immunoglobulin secretion, and angiogenesis. Genetic polymorphisms in the IL-6 gene could explain regional predispositions, such as the higher incidence in Asia. Environmental factors like sunlight exposure and potential infectious triggers have been hypothesized, though not definitively proven. Importantly, the plasma cell infiltrate is polyclonal, distinguishing it from monoclonal proliferations seen in plasma cell neoplasms like multiple myeloma or cutaneous plasmacytoma.
Clinical features
Patients typically present with a characteristic triad: persistent cutaneous lesions, polyclonal hypergammaglobulinemia, and regional lymphadenopathy. Skin manifestations include multiple round-to-oval, red-to-dark-brown macules, papules, nodules, and plaques, most commonly on the trunk but also involving the face, neck, axillae, and extremities. Lesions are often pruritic, indurated, and may show central hair loss or exudation in severe cases.
Systemic symptoms occur in many cases, including anaemia, fever, fatigue, and elevated erythrocyte sedimentation rate (ESR). Extracutaneous involvement, such as superficial lymphadenopathy, is frequent, and rare cases report ocular or other organ infiltration. Laboratory findings commonly reveal polyclonal hypergammaglobulinaemia, particularly elevated IgG and sometimes IgG4, without light chain restriction. The condition follows a chronic, benign course, with potential for partial spontaneous remission or progression to more widespread involvement.
- Key clinical sites: Trunk (most common), face, neck, axillae, extremities.
- Lesion morphology: Reddish-brown macules, papules, plaques, nodules; often symmetric and persistent.
- Associated findings: Polyclonal hypergammaglobulinaemia, anaemia, lymphadenopathy, elevated ESR.
Pathology
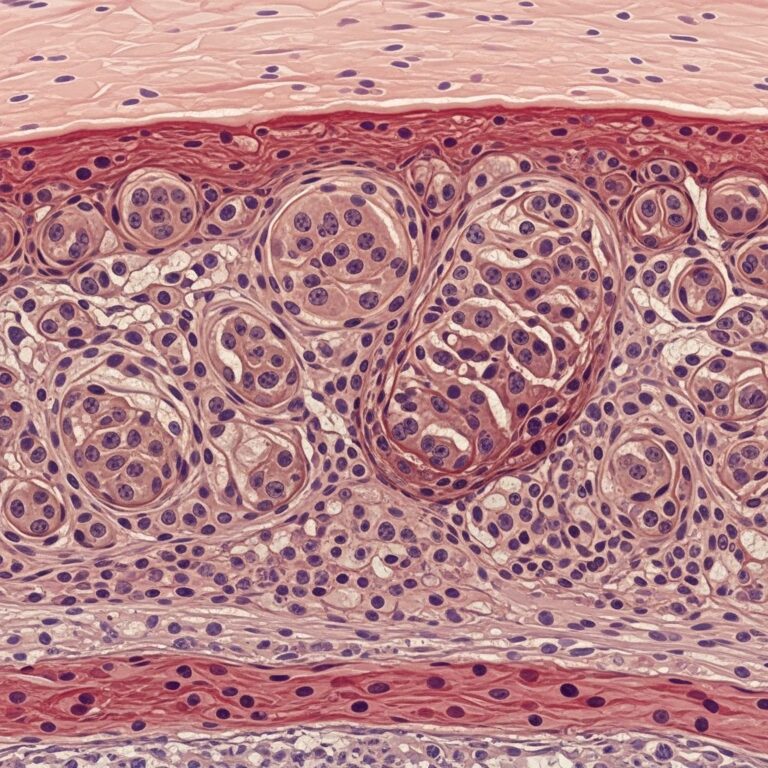
Histopathological examination is pivotal for diagnosis, revealing a moderately dense perivascular and interstitial infiltrate of mature plasma cells throughout the superficial and deep dermis. The epidermis is typically unremarkable, without significant acanthosis, spongiosis, or interface changes. Plasma cells predominate, admixed with lymphocytes, histiocytes, and occasional eosinophils or mast cells. Lymphoid follicles with reactive germinal centres may be present, mimicking lymphoplasmacytic processes.
High magnification shows characteristic plasma cell morphology: eccentric nuclei, clock-face chromatin, and perinuclear hof. Immunohistochemistry confirms polyclonality with equal kappa and lambda light chain expression, negative for light chain restriction. Additional markers include CD138+, CD79a+, MUM1+, and variable IgG4 positivity, raising considerations of IgG4-related disease overlap. There is no significant atypia, mitoses, or Dutcher bodies, which would suggest malignancy.
In lymph node biopsies, if performed, similar polyclonal plasma cell aggregates are seen in paracortical areas, without effacement of architecture.
Microscopic (histologic) description
- Dense perivascular and interstitial dermal infiltrate of mature plasma cells
- Extension into superficial and deep dermis
- Admixture of small lymphocytes, histiocytes, mast cells
- Occasional lymphoid aggregates with germinal centres
- No epidermal involvement or atypia
Microscopic (histologic) images
Typical low-power view demonstrates sheets of plasma cells filling the dermis (H&E). Higher power highlights mature plasma cells with eccentric nuclei and perinuclear clearing.
Histopathology — skin
The hallmark is a top-heavy to diffuse infiltrate of mature plasma cells in perivascular distribution, extending from papillary to reticular dermis. Fibrosis may be present in chronic lesions, and vascular proliferation can occur due to angiogenic factors. Special stains (e.g., Congo red) are negative for amyloid, and no Russell bodies are typically noted unless secondary changes are present.
| Feature | Description |
|---|---|
| Infiltrate pattern | Perivascular, interstitial, nodular |
| Cell type | Mature plasma cells (predominant) |
| Depth | Superficial and deep dermis |
| Immunohistochemistry | Polyclonal (kappa:lambda equal), CD138+ |
| Other cells | Lymphocytes, histiocytes, eosinophils, mast cells |
Histopathology — lymph node
When biopsied, lymph nodes show paracortical expansion by polyclonal plasma cells, preserving nodal architecture. No monocytoid B-cells or sheets of immunoblasts.
Immunohistochemistry and special stains
- Plasma cells: CD79a+, CD138+, MUM1+, IRF4+; polyclonal kappa/lambda
- Lymphocytes: CD3+, CD20+ (polyclonal)
- Negative: CD5, CD10, CD23 (to exclude lymphoma); light chain restriction
- Special stains: Negative for HHV-8, EBV; Gram, AFB, GMS negative
Differential diagnosis
The key challenge is distinguishing benign polyclonal plasmacytosis from malignant or secondary causes:
| Differential | Key Distinguishing Features |
|---|---|
| Plasma cell neoplasm (plasmacytoma) | Monoclonal light chain restriction, atypia, Dutcher bodies |
| Multiple myeloma | Bone marrow involvement, lytic lesions, monoclonal paraprotein |
| Lymphoplasmacytic lymphoma | Monoclonal B-cells, MYD88 mutation, bone marrow disease |
| Secondary plasmacytosis (e.g., syphilis, HIV, lupus) | Positive serology, systemic autoimmune features, regression with treatment |
| IgG4-related disease | High IgG4/IgG ratio (>40%), storiform fibrosis, obliterative phlebitis |
| Pseudolymphoma | Predominantly lymphocytic, atypical follicles, drug history |
Diagnosis
Diagnosis requires clinicopathologic correlation: characteristic lesions, histopathology showing polyclonal plasma cells, negative workup for malignancy/systemic plasma cell dyscrasia (serum protein electrophoresis, bone marrow biopsy if indicated), and exclusion of infections (HIV, syphilis, etc.). Polyclonal hypergammaglobulinaemia supports the diagnosis.
Treatment
No standardized therapy exists due to rarity and benign course. Options include:
- Observation: For asymptomatic or mild cases
- Topical/intralesional steroids: For localized plaques
- Phototherapy (PUVA, narrowband UVB): Effective partial remission in several reports
- Systemic steroids: Low-dose prednisone (e.g., 20 mg/day) for widespread disease
- Other: Thalidomide, rituximab, cyclosporine (anecdotal)
Spontaneous regression occurs rarely; monitor for malignant transformation, though exceptional.
Frequently asked questions
What is cutaneous plasmacytosis?
A rare benign proliferation of polyclonal mature plasma cells in skin, with possible systemic features like hypergammaglobulinaemia and lymphadenopathy.
Is cutaneous plasmacytosis malignant?
No, it is reactive/benign; plasma cells are polyclonal without atypia or progression to myeloma reported commonly.
How is the diagnosis confirmed?
By skin biopsy showing dense mature plasma cell infiltrate with immunohistochemistry proving polyclonality (equal kappa/lambda).
What are the treatment options?
Observation, topical/systemic steroids, phototherapy; no curative therapy, management is symptomatic.
Does it affect specific populations?
Predominantly Asians (Japanese), but reported globally; chronic, non-fatal.
References
- Primary cutaneous plasmacytosis — Orphanet. 2023. https://www.orpha.net/en/disease/detail/451602
- Cutaneous plasmacytosis: A rare entity with unique presentation — Indian Journal of Dermatology, Venereology and Leprology. 2018-05-01. https://ijdvl.com/cutaneous-plasmacytosis-a-rare-entity-with-unique-presentation/
- Cutaneous Plasmacytosis: A Clinicopathologic Study — PubMed (Annals of Dermatology). 2017-04-07. https://pubmed.ncbi.nlm.nih.gov/28475511/
- Cutaneous plasmacytosis Characterized by Head Plaques — PMC (Indian Journal of Dermatology). 2023. https://pmc.ncbi.nlm.nih.gov/articles/PMC10408662/
- Cutaneous and systemic plasmacytosis — VisualDx. 2025 (updated). https://www.visualdx.com/visualdx/diagnosis/cutaneous+and+systemic+plasmacytosis?diagnosisId=57077&moduleId=101
Similar Articles






Read full bio of medha deb