Dermatofibrosarcoma Protuberans: Types, Diagnosis, Treatment
Understanding DFSP: A rare skin sarcoma with genetic origins and multidisciplinary treatment approaches.

Dermatofibrosarcoma Protuberans: An Overview
Dermatofibrosarcoma protuberans (DFSP) is a rare proliferative skin tumor classified as a soft-tissue sarcoma that arises from the deeper layers of skin. This locally aggressive neoplasm is characterized by a unique genetic alteration and a distinctive clinical presentation. While DFSP has a high propensity for local recurrence, metastatic disease remains exceptionally rare, occurring in fewer than 5% of cases historically, though more recent literature documents slightly higher rates. Understanding this condition is essential for dermatologists, surgeons, and patients, as early recognition and appropriate management significantly impact outcomes.
Clinical Presentation and Characteristics
DFSP typically presents as a slow-growing skin lesion that evolves over months to years. The characteristic appearance varies depending on the disease stage and specific variant.
Initial Presentation
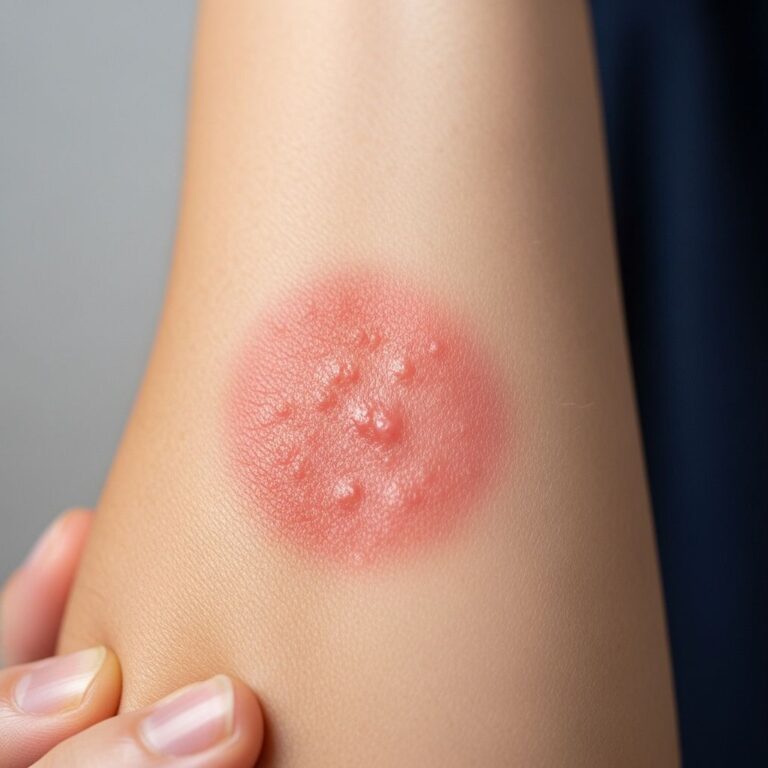
The tumor most often begins as a small, firm patch of skin, usually measuring 1 to 5 centimeters in diameter. The lesion is typically purplish, reddish, or flesh-colored in appearance. In its early stages, DFSP may present as a flat or depressed patch (plaque) rather than an elevated nodule, particularly in the preprotuberant phase. This preprotuberant stage can persist for a mean duration of approximately 7 years before the lesion becomes visibly bulging and nodular.
Progressive Features
As the tumor progresses, it gradually transforms into a raised nodule that may become increasingly firm and well-demarcated. The lesion continues its characteristic slow growth pattern. Importantly, the tumor extends beyond its clinically apparent surface into surrounding tissues, with irregular and frequently deep subclinical extensions that are not visibly apparent to the naked eye. This feature is critical for treatment planning, as complete removal requires recognition of these deep extensions.
Genetic and Pathologic Basis
Molecular Characteristics
The genetic foundation of DFSP is a reciprocal translocation t(17;22)(q22;q13) that produces a pathognomonic COL1A1-PDGFB gene fusion. This chromosomal abnormality results in fusion of the collagen type I alpha 1 gene (COL1A1; located on 17q22) with the platelet-derived growth factor beta gene (PDGFβ; located on 22q13). This fusion protein drives the neoplastic proliferation characteristic of DFSP.
Histopathologic Features
Microscopically, DFSP demonstrates distinctive architectural and cytologic patterns:
- Spindled cells arranged in storiform (pinwheel) or fascicular patterns
- Typically bland cytomorphology with minimal cytologic atypia
- Variable cellularity ranging from moderately cellular to highly cellular lesions
- Positive CD34 immunohistochemical staining, which is present in the majority of cases
- Negative factor XIIIa staining, helping distinguish DFSP from dermatofibroma
Fibrosarcomatous Transformation
A significant pathologic variant is fibrosarcomatous transformation (FS-DFSP), which occurs in approximately 5-15% of cases. This transformation is characterized by:
- Increased cellularity and cytologic atypia
- Elevated mitotic activity (greater than 5 mitoses per 10 high-power fields)
- Loss or marked reduction in CD34 immunostaining with negative staining patterns
- More aggressive biological behavior and higher metastatic potential
For equivocal lesions, confirmatory testing with fluorescence in situ hybridization (FISH), polymerase chain reaction (PCR), or conventional cytogenetics may be employed to demonstrate the COL1A1-PDGFB translocation.
Diagnostic Approach
Clinical Examination
The diagnostic process begins with careful visual inspection and palpation of the skin lesion. Healthcare providers assess the size, color, texture, and borders of the lesion. The clinical history, including duration and growth pattern, is particularly important given DFSP’s characteristically slow growth.
Skin Biopsy
A tissue biopsy is essential for definitive diagnosis. The healthcare professional removes a small amount of tissue, which is then examined under microscopy. Laboratory analysis identifies the characteristic histopathologic features and immunophenotypic profile. A skin biopsy is the standard method for confirming DFSP and excluding other differential diagnoses.
Imaging Studies
Advanced imaging is sometimes employed to assess the extent of disease and guide treatment planning. Magnetic resonance imaging (MRI) can delineate the tumor’s deep extensions and relationship to surrounding structures. In some cases, additional imaging may be warranted to evaluate for regional lymph node involvement or distant metastases, particularly in fibrosarcomatous variants.
Treatment Principles
Surgical Excision: The Gold Standard
Surgery is the primary and most effective treatment for DFSP. Because of the tumor’s proclivity for irregular and frequently deep subclinical extensions, meticulous surgical planning is essential to ensure complete removal at the time of initial therapy.
Mohs Micrographic Surgery
Mohs micrographic surgery (MMS) is widely recognized as an optimal surgical approach for DFSP. This technique offers two critical advantages:
- Ensures complete removal of the tumor by allowing real-time assessment of surgical margins using frozen-section microscopy
- Maximizes tissue preservation, which is particularly valuable for lesions in cosmetically or functionally sensitive areas
Mohs surgery involves systematic layer-by-layer excision with immediate histopathologic examination of all margins, allowing surgeons to achieve clear margins while removing minimal normal tissue.
Wide Surgical Excision
When Mohs micrographic surgery is not available, wide excision with at least 2-centimeter margins to the investing fascia of muscle or pericranium is recommended. This approach should achieve clear pathologic margins when clinically feasible. The surgical plan must account for the tumor’s size, anatomic location, and cosmetic considerations.
Management of Surgical Margins
The status of surgical margins is critical for determining adjuvant treatment and prognosis:
- Negative margins: When clear margins are achieved, observation without adjuvant therapy is typically recommended
- Positive margins: Re-resection is recommended whenever possible, with the goal of achieving clear margins
- Indeterminate margins: If uncertainty exists regarding margin clearance, split-thickness skin grafting (STSG) may be considered to facilitate monitoring for recurrence
Comprehensive, meticulous en face permanent section examination of all surgical margins is essential to accurately determine margin status.
Reconstruction
Following tumor removal, reconstructive surgery may be necessary to restore form and function. However, extensive reconstruction involving tissue undermining or significant tissue movement should be avoided or delayed until negative histologic margins are confirmed. This precaution prevents potential tumor seeding if margins prove positive upon final pathologic examination.
Adjuvant and Systemic Therapy
Radiation Therapy
Adjuvant radiation therapy (RT) is recommended for positive or indeterminate margins. Standard dosing includes:
- 50-60 Gray (Gy) for indeterminate or positive margins
- Up to 66 Gy for overtly positive margins or gross residual tumor
- Radiation fields should extend widely beyond the surgical margin (typically 3-5 cm when clinically feasible)
In contrast, radiation is not recommended for patients with negative surgical margins. For recurrent or metastatic disease where further surgical resection is not feasible and radiation has not been previously administered, RT remains a valuable treatment option.
Targeted Therapy with Imatinib Mesylate
Imatinib mesylate (Gleevec) is a tyrosine kinase inhibitor that has emerged as an important systemic therapy option for DFSP. This targeted therapy medicine attacks the specific proteins produced by cancer cells containing the COL1A1-PDGFB fusion, causing cancer cell death. Imatinib is considered in the following clinical scenarios:
- Unresectable disease when surgery is not a viable option
- Neoadjuvant therapy prior to surgery to potentially downstage tumors and improve surgical outcomes
- Recurrent disease after initial surgical treatment
- Metastatic disease as part of multidisciplinary treatment
Chemotherapy
For rare cases of metastatic disease, various chemotherapy regimens commonly used for soft-tissue sarcomas may be considered. These include:
- AIM regimen (doxorubicin/ifosfamide/mesna)
- Single-agent doxorubicin
- Single-agent ifosfamide
The selection of chemotherapy is individualized and typically coordinated through multidisciplinary consultation.
Treatment of Recurrent Disease
Local recurrence remains a significant challenge in DFSP management, with recurrence rates reaching up to 55% after initial excision if inadequate surgical margins are achieved. Management of recurrent DFSP includes:
- Re-resection with meticulous surgical technique to achieve negative margins
- Radiation therapy if not previously administered
- Imatinib mesylate, particularly when further surgery would result in unacceptable functional or cosmetic morbidity
- Multidisciplinary consultation to coordinate comprehensive treatment strategies
Metastatic Disease
True metastatic DFSP is uncommon, developing in fewer than 5% of traditionally reported cases. However, metastatic spread can occur via both lymphatic and hematogenous routes. Management of metastatic disease requires multidisciplinary consultation to coordinate treatment, which may include:
- Clinical trials testing novel therapies
- Imatinib mesylate or other targeted agents
- Systemic chemotherapy
- Radiation therapy
- Re-resection of resectable metastatic sites when feasible
Follow-Up and Surveillance
Close follow-up is essential given the risk of local recurrence. Patients should undergo regular clinical examination of the surgical site and surrounding skin. The frequency of follow-up visits should be individualized based on margin status, tumor characteristics, and treatment received. Long-term surveillance is recommended to detect early signs of recurrence or metastatic disease.
Prognosis and Outcomes
The prognosis for DFSP is generally favorable when appropriate surgical treatment is provided. Most patients with negative margins and standard-type DFSP (without fibrosarcomatous transformation) have excellent long-term survival. However, the high risk of local recurrence necessitates meticulous surgical technique and close follow-up. Fibrosarcomatous variants carry a more guarded prognosis due to their increased metastatic potential.
Frequently Asked Questions
Q: How long does it take for DFSP to develop?
A: DFSP grows slowly over months to years. The preprotuberant phase (flat or depressed lesion) can persist for approximately 7 years on average before the lesion becomes noticeably elevated and nodular. Early recognition and treatment can prevent progression to advanced disease.
Q: Is DFSP a form of skin cancer?
A: Yes, DFSP is classified as a soft-tissue sarcoma arising from the deeper layers of skin. It is a malignant tumor, though metastatic disease is rare when properly treated with complete surgical excision.
Q: Can DFSP be treated without surgery?
A: Surgery is the gold standard and most effective treatment. However, for patients with unresectable disease or those who cannot tolerate surgery, imatinib mesylate or radiation therapy may be used as alternative or adjunctive treatments.
Q: What is the recurrence rate for DFSP?
A: Local recurrence rates can reach up to 55% after initial excision if inadequate margins are achieved. This emphasizes the importance of complete surgical removal with clear margins and appropriate follow-up.
Q: What does the COL1A1-PDGFB translocation mean?
A: This is a characteristic chromosomal abnormality (t(17;22)) found in DFSP that creates a fusion gene driving tumor growth. This translocation is diagnostic and is also the target of imatinib therapy, making it clinically significant for treatment planning.
References
- Dermatofibrosarcoma Protuberans: An Updated Review — PubMed Central. 2024. https://pmc.ncbi.nlm.nih.gov/articles/PMC11430793/
- Dermatofibrosarcoma protuberans – Diagnosis and treatment — Mayo Clinic. 2024. https://www.mayoclinic.org/diseases-conditions/dermatofibrosarcoma-protuberans/diagnosis-treatment/drc-20576979
- NCCN Guidelines Version 1.2019: Dermatofibrosarcoma Protuberans Treatment — National Comprehensive Cancer Network. 2019. http://medi-guide.meditool.cn/ymtpdf/5D967832-C0EB-D179-57EC-1F241F8D2A55.pdf
- Dermatofibrosarcoma protuberans – Genetics — MedlinePlus. 2024. https://medlineplus.gov/genetics/condition/dermatofibrosarcoma-protuberans/
- Dermatofibrosarcoma Protuberans: A Guide for Medical Students — MedEdPage. 2022. https://www.youtube.com/watch?v=fdWzWpREu2I
Similar Articles






Read full bio of medha deb