Dystrophic Epidermolysis Bullosa: 7 Essential Insights
Rare genetic skin disorder causing extreme fragility, blistering, and scarring from minor friction or trauma.

Dystrophic epidermolysis bullosa is one of the four main types of epidermolysis bullosa, a group of rare genetic skin disorders characterised by fragile skin that blisters easily. Dystrophic epidermolysis bullosa (DEB, also called dermolytic epidermolysis bullosa or epidermolysis bullosa dystrophica) affects approximately 25% of all epidermolysis bullosa cases and results from mutations in the COL7A1 gene encoding type VII collagen, leading to separation within the dermal layer below the basement membrane.
What is dystrophic epidermolysis bullosa?
Dystrophic epidermolysis bullosa is inherited in either an autosomal dominant or recessive manner. Recessive forms are generally more severe, with widespread blistering from birth, while dominant forms tend to be milder and localised. The hallmark is extreme skin fragility due to defective anchoring fibrils, causing blisters from minor friction that heal with scarring, milia formation, and potential mutilating deformities.
- Blisters form subepidermally within the papillary dermis.
- Healing leads to atrophic scars, nail dystrophy, and mucous membrane involvement.
- Severe cases risk squamous cell carcinoma (SCC), contractures, and reduced life expectancy.
Who gets dystrophic epidermolysis bullosa?
Dystrophic epidermolysis bullosa affects all ethnic groups equally and has an estimated prevalence of 3 per million worldwide. It manifests at birth or early infancy, with recessive severe generalised (RDEB-sev gen) being the most debilitating form.
- Incidence: Approximately 2.6 per million live births for recessive DEB.
- Dominant forms are rarer and milder.
What causes dystrophic epidermolysis bullosa?
DEB arises from pathogenic variants in the COL7A1 gene on chromosome 9p21.3, which codes for type VII collagen – the main component of anchoring fibrils that secure the epidermis to the dermis. Over 1,000 mutations are known, with glycine substitutions in the triple helical domain common in dominant DEB, and nonsense or frameshift mutations causing absent protein in recessive forms.
Autosomal dominant DEB results from heterozygous mutations producing partially functional collagen. Autosomal recessive DEB requires biallelic mutations, leading to complete absence or severe dysfunction of type VII collagen.
What are the clinical features of dystrophic epidermolysis bullosa?
Clinical severity correlates with residual type VII collagen function. Blisters and erosions occur from minor trauma, healing with white atrophic scars, milia (small cysts), and nail loss. Mucosal involvement affects 80% of severe cases.
- Skin: Tense blisters on extremities, trunk; progress to ulcers, scarring, pseudosyndactyly (toes > fingers).
- Nails: Dystrophic, absent in 90% severe cases.
- Mucosa: Oral blisters, microstomia, dental caries, oesophageal strictures (causing dysphagia, malnutrition).
- Other: Anaemia, growth failure, pruritus, constipation.
Severe generalised recessive subtype
RDEB-sev gen presents with widespread erosions at birth, often with aplasia cutis. Profound scarring causes mitten deformities, joint contractures, and oesophageal stenosis by adolescence.
Other subtypes
| Subtype | Inheritance | Key Features |
|---|---|---|
| Severe generalised (RDEB-sev gen) | Recessive | Widespread blisters, albopapuloid lesions, SCC risk >90% by age 45. |
| Generalised non-severe (RDEB-gen non-sev) | Recessive | Hand/foot predominance, milder scarring. |
| Localised (RDEB-loc) | Recessive | Blisters limited to trauma sites. |
| Dominant (DDEB) | Dominant | Milder; nail dystrophy, albopapuloid lesions. |
| Pruriginosa | D/R | Itchy papules, lichenoid changes. |
| Inversa | Recessive | Flexural predominance. |
| Bullous dermolysis of newborn | Dominant | Neonatal blisters resolve by age 2. |
Diagnosis of dystrophic epidermolysis bullosa
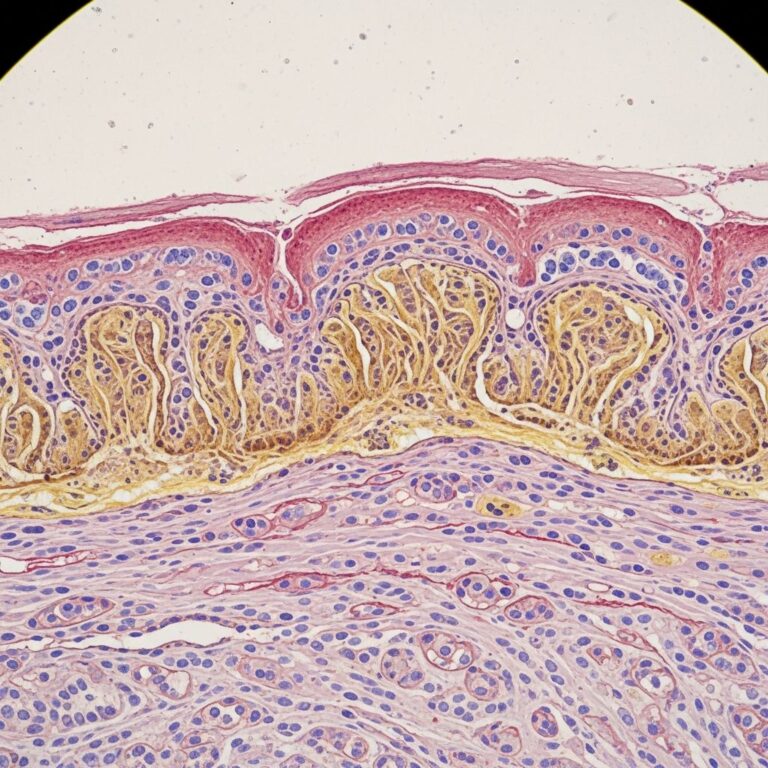
Suspected clinically in neonates with unexplained blisters. Confirmed by skin biopsy showing sub-lamina densa cleft on transmission electron microscopy (TEM) and reduced type VII collagen on immunofluorescence.
- IFM: Absent/reduced COL7 staining at basement membrane.
- TEM: Hypoplastic/rudimentary anchoring fibrils.
- Genetic testing: COL7A1 sequencing (gold standard).
How is dystrophia epidermolysis bullosa treated?
No cure exists; management is supportive, multidisciplinary, focusing on wound care, pain control, nutrition, and complication prevention.
- Wound care: Non-adherent dressings (e.g., Mepilex), silicone products; infection surveillance.
- Pain: WHO ladder analgesics, gabapentin for neuropathic pain.
- Nutrition: High-calorie feeds, oesophageal dilation.
- Surgery: Syndactyly release (every 2-3 years), SCC excision.
- Emerging: Gene therapy (e.g., beremagene geperpavec – topical COL7A1), cell therapy.
What is the outcome for dystrophic epidermolysis bullosa?
Varies by subtype. Dominant/mild recessive: Normal lifespan. Severe RDEB: Frequent hospitalisations, wheelchair dependence by teens, SCC mortality (cumulative risk 78-90%). Multidisciplinary care improves quality of life.
Prevention of complications in dystrophic epidermolysis bullosa
- Daily wound inspection, infection prophylaxis.
- Padding trauma-prone areas, protective clothing.
- Regular dental/ophthalmic review.
- Vaccinations, iron supplementation for anaemia.
- Skin surveillance for SCC from adolescence.
Recent advances in dystrophic epidermolysis bullosa
Vyjuvek® (beremagene geperpavec-svdt) – FDA-approved 2023 topical gene therapy for RDEB wounds, delivering functional COL7A1 via HSV-1 vector, improving healing. Ongoing trials: Stem cell therapy, anti-inflammatory biologics.
Frequently asked questions
What is the most severe form of dystrophic epidermolysis bullosa?
Severe generalised recessive DEB (Hallopeau-Siemens type), with complete type VII collagen loss, widespread scarring, and high SCC risk.
Does dystrophic epidermolysis bullosa affect internal organs?
Yes, oesophagus (strictures), mouth, anus; rarely bladder, respiratory tract.
Can dystrophic epidermolysis bullosa be cured?
Not currently, but supportive care and emerging gene therapies significantly improve outcomes.
What is the cancer risk in DEB?
Lifetime SCC risk up to 90% in severe RDEB; chronic wounds are precursors.
How is DEB inherited?
Autosomal dominant (milder) or recessive (severe); genetic counselling recommended.
References
- Dystrophic epidermolysis bullosa — MedlinePlus Genetics, National Library of Medicine. 2023-06-01. https://medlineplus.gov/genetics/condition/dystrophic-epidermolysis-bullosa/
- Epidermolysis Bullosa — StatPearls, NCBI Bookshelf, National Center for Biotechnology Information. 2024-07-17. https://www.ncbi.nlm.nih.gov/books/NBK599531/
- Dystrophic EB — DEBRA of America. 2024. https://www.debra.org/understanding-eb/dystrophic-eb
Similar Articles






Read full bio of medha deb