Familial Adenomatous Polyposis (FAP): Causes, Diagnosis, and Treatment
Understanding FAP: A hereditary syndrome with nearly 100% colorectal cancer risk requiring early intervention.
: Causes, Diagnosis, and Treatment")
Understanding Familial Adenomatous Polyposis (FAP)
Familial adenomatous polyposis (FAP) is a hereditary syndrome that significantly increases your risk of developing colorectal cancer. Unlike the general population where colorectal cancer develops sporadically, individuals with FAP face a lifetime cancer risk of nearly 100%. This condition represents a critical medical challenge that demands early detection and proactive management to prevent life-threatening complications.
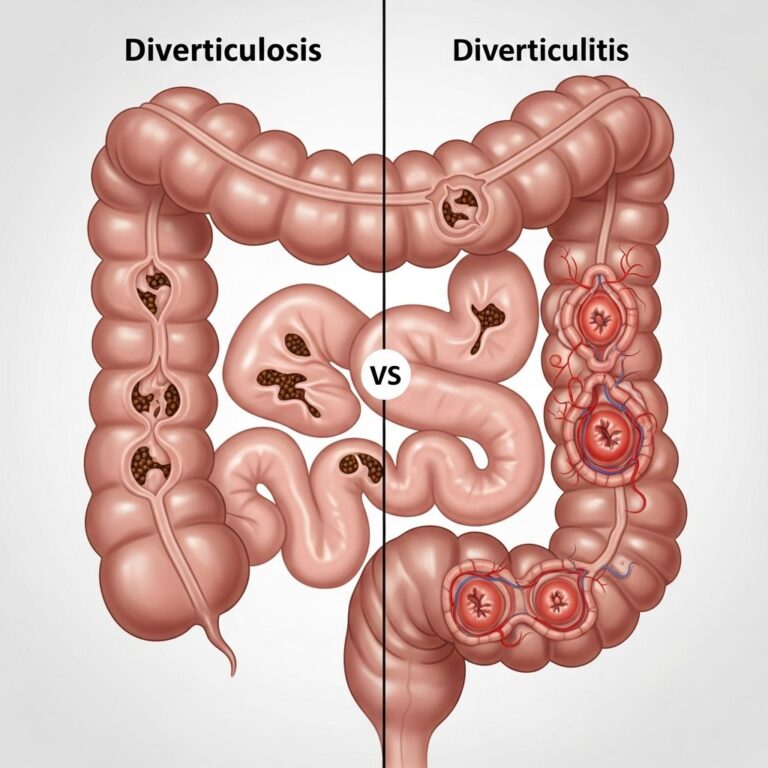
FAP is characterized by the development of hundreds to thousands of adenomatous polyps throughout the colon, typically beginning in the teenage years or early adulthood. The sheer number of these growths creates an overwhelming statistical probability that at least one will become cancerous if left untreated. Without intervention, most people with FAP will develop colorectal cancer by their middle-age years, making it one of the most aggressive hereditary cancer syndromes.
What Is Familial Adenomatous Polyposis?
FAP is an inherited genetic disorder that predisposes affected individuals to develop multiple colon polyps, which are precancerous growths. The condition typically manifests with the appearance of at least 100 polyps in the colon, though some patients develop significantly more. The accumulation of these polyps creates an environment where malignant transformation becomes virtually inevitable without surgical intervention.
The polyps in FAP grow more frequently and at a faster rate than polyps in individuals without the syndrome. This accelerated polyp formation dramatically increases the likelihood that at least one polyp will develop into cancer. The condition accounts for approximately 0.5% of all colorectal cancer cases, making it relatively rare but extremely significant for affected families.
Prevalence and Incidence
Familial adenomatous polyposis is estimated to affect approximately 1 in 8,000 people worldwide. While this may seem uncommon, the condition has profound implications for affected families. Several subtypes of FAP exist, including attenuated FAP (AFAP), Gardner syndrome, and Turcot syndrome. These variants represent between 5% and 10% of all familial adenomatous polyposis cases and typically present with fewer polyps or additional extracolonic features.
The Cleveland Clinic’s Hereditary Colorectal Cancer Registry, established in 1979, represents the largest single-institution registry for FAP in the world. This comprehensive registry has served over 1,000 families with FAP, providing crucial data on disease progression, treatment outcomes, and long-term patient management.
Genetic Basis of FAP
The APC Gene Mutation
Familial adenomatous polyposis is caused by a mutation in the adenomatous polyposis coli gene, commonly referred to as the APC gene. This gene serves as a tumor suppressor, meaning its normal function is to prevent cells from growing out of control and forming tumors. When the APC gene is mutated, it loses this protective function, allowing cells in the colon to proliferate abnormally and form numerous polyps.
The APC gene mutation associated with FAP is a germline mutation, occurring at conception rather than developing during a person’s lifetime. This is why the condition runs in families—the mutation is present in every cell of an affected individual’s body from birth.
Inheritance Pattern
FAP follows an autosomal dominant inheritance pattern, which means that if one of your parents has the mutation, you have a 50% chance of inheriting it. However, an important distinction exists: approximately 30% of FAP cases result from spontaneous mutations that occur without a family history of the condition. This means that about 30% of newly diagnosed FAP patients have no affected relatives, and they are the first in their families to carry the mutation.
For individuals with a family history of FAP, genetic counseling and testing are crucial steps in understanding personal cancer risk and developing an appropriate surveillance and management plan.
Clinical Features and Symptoms
Polyp Development
Colon polyps in FAP typically begin to appear much earlier than in the general population, often during the teenage years or early twenties. Many individuals with FAP will develop hundreds or even thousands of polyps throughout their colon. Those with the attenuated form (AFAP) develop at least 20 polyps, which is still significantly more than the average person.
These polyps grow rapidly and frequently compared to sporadic polyps that develop in individuals without hereditary syndromes. The accelerated growth rate and sheer abundance of polyps make FAP particularly dangerous for colorectal cancer development.
Symptoms
In many cases, individuals with FAP do not experience symptoms until polyps have grown large enough to cause complications. Early detection through screening rather than symptom recognition is therefore essential. When symptoms do occur, they may include:
- Rectal bleeding or blood in the stool
- Diarrhea or changes in bowel habits
- Chronic abdominal pain or cramping
- Anemia (from chronic blood loss)
The greater number and faster growth of polyps in FAP compared to sporadic polyps make them more likely to cause noticeable symptoms. However, many patients discover their condition through screening colonoscopies rather than clinical symptoms.
Associated Cancers
Beyond colorectal cancer, individuals with FAP have an increased risk of developing other malignancies. These include gastric cancer, small intestine cancer, pancreatic cancer, biliary tract cancer, and thyroid cancer. Additionally, people with FAP may develop other abnormal growths in the skin, soft tissues, teeth, and bones, as well as noncancerous conditions like desmoid tumors.
Even after colectomy surgery to remove the colon, patients require lifelong surveillance for these extracolonic manifestations and may require additional procedures to manage associated tumors.
Diagnosis of Familial Adenomatous Polyposis
Genetic Testing
Diagnosis of FAP begins with genetic testing for the APC gene mutation. A genetic test involves collecting a DNA sample through blood or saliva and analyzing it for specific gene mutations. Identifying the presence of an APC mutation is the cornerstone of FAP diagnosis and allows for precise risk stratification and surveillance planning.
Diagnostic Criteria
Formal diagnosis of FAP is established through specific clinical and genetic criteria. A diagnosis is confirmed when an individual has either at least 100 adenomatous polyps in the colon combined with an identified APC gene mutation, or 20 or more polyps for the attenuated form (AFAP) with genetic confirmation.
Colonoscopy Screening
Once genetic testing confirms an APC mutation, colonoscopy screening is the next critical diagnostic step to identify and characterize the adenomatous polyps present. Children in families known to be affected by FAP should begin yearly colonoscopy screenings at age 10 to detect polyp development early.
Regular colonoscopy allows healthcare providers to track the number, size, and appearance of polyps, which helps guide treatment decisions and timing of surgery. The colonoscopy can also be therapeutic, allowing removal of individual polyps when their number is still manageable.
Treatment and Management Strategies
Initial Management
Treatment for FAP involves a combination of lifelong surveillance and eventual surgical intervention. In the early stages, if an individual has fewer polyps or is diagnosed with AFAP, healthcare providers may attempt to remove polyps individually during colonoscopy procedures, a technique called polypectomy. This approach allows temporary management of polyp burden while delaying the need for major surgery.
Recent advances in medical therapy show promise. Combination therapy approaches have demonstrated the ability to delay the need for lower gastrointestinal surgery in some patients with FAP, offering patients additional years of management without colectomy.
Surgical Intervention
Surgery becomes necessary when there are too many polyps to manage through colonoscopic removal or when polyps show signs of malignant transformation. Most people with FAP will eventually require complete removal of the colon, a procedure called total colectomy, sometime during their early adult years. Some individuals also require removal of the rectum (proctocolectomy) depending on polyp distribution and clinical presentation.
Surgical management of FAP is complex and requires both sound judgment and technical skills from experienced surgeons. Advanced surgical techniques including minimally invasive and robotic surgery, as well as pancreas-sparing duodenectomy for managing upper gastrointestinal polyps, are now available at specialized centers.
Specialized Care and Multidisciplinary Approach
Comprehensive management of FAP requires expertise and coordination among multiple medical specialties. The Cleveland Clinic’s Hereditary Colorectal Cancer Registry exemplifies this multidisciplinary approach, involving colorectal surgeons, gastroenterologists, genetic counselors, and mental health professionals working together to optimize patient outcomes.
Patients with FAP benefit significantly from care at specialized registries or through healthcare providers with extensive experience managing hereditary colorectal cancer syndromes. Such centers employ advanced endoscopic techniques, offer genetic counseling, and coordinate comprehensive surveillance protocols to ensure early detection and optimal management.
Frequently Asked Questions
Q: What is the difference between FAP and sporadic colon polyps?
A: Sporadic colon polyps develop randomly as people age and typically occur in limited numbers. FAP, by contrast, causes hundreds to thousands of polyps beginning in teenage years due to an inherited genetic mutation. The abundance and accelerated growth of polyps in FAP make cancer development virtually inevitable without intervention.
Q: If I have FAP, will I definitely develop colon cancer?
A: Without treatment, the risk of developing colorectal cancer with familial adenomatous polyposis approaches 100%. However, with appropriate surveillance and preventive surgery, cancer development can be prevented. This is why early detection and management are so critical.
Q: At what age should screening begin for children with a family history of FAP?
A: Children in families known to be affected by FAP should begin yearly colonoscopy screenings at age 10. Early screening allows detection of polyps as they develop and guides decisions about management and timing of potential surgery.
Q: Can FAP be managed without surgery?
A: In early stages with fewer polyps or in AFAP, colonoscopic removal of individual polyps may be attempted. However, as polyp burden increases, surgery typically becomes necessary. Recent medical therapies show promise in delaying surgery, but most people with FAP will eventually require colectomy.
Q: Are there other conditions similar to FAP?
A: Yes, MUTYH-associated polyposis (MAP) is a similar hereditary syndrome that causes adenomatous colon polyps but is caused by a different gene mutation in the MUTYH gene rather than the APC gene. Other hereditary colorectal cancer syndromes include Lynch syndrome and various hamartomatous polyposis syndromes.
Q: What happens after colectomy surgery for FAP?
A: Even after colectomy, individuals with FAP require lifelong surveillance for other cancers and abnormal growths that can develop in various organs. They may need additional screening and procedures to manage extracolonic manifestations like desmoid tumors or upper gastrointestinal polyps.
References
- Familial Adenomatous Polyposis (FAP) — Cleveland Clinic. 2024. https://my.clevelandclinic.org/health/diseases/16993-familial-adenomatous-polyposis-fap
- Combination Therapy Delays Need for Lower GI Surgery in Patients with Familial Adenomatous Polyposis — Consult QD, Cleveland Clinic. 2024. https://consultqd.clevelandclinic.org/tag/familial-adenomatous-polyposis
- Hereditary Colorectal Cancer Registry: A Multidisciplinary Approach to Familial Adenomatous Polyposis and Lynch Syndrome — Revista Médica Clínica Las Condes / Cleveland Clinic. 2016. https://www.elsevier.es/es-revista-revista-medica-clinica-las-condes-202-articulo-hereditary-colorectal-cancer-registry-a-S0716864017300913
- Spontaneous Mutation in Familial Adenomatous Polyposis — Rustin R, et al. PubMed/NCBI. 1990. https://pubmed.ncbi.nlm.nih.gov/2153067/
- Familial Adenomatous Polyposis: Challenges and Pitfalls of Surgical Management — Warrier SK, et al. PubMed/NCBI. 2012. https://pubmed.ncbi.nlm.nih.gov/23730222/
Similar Articles






Read full bio of medha deb