Familial Benign Pemphigus Pathology: Clinical Guide
Detailed pathology of Hailey-Hailey disease, a genetic blistering disorder with acantholysis in intertriginous areas.

Next review due: July 2027
Introduction
Familial benign pemphigus, also known as Hailey-Hailey disease (HHD), is a rare autosomal dominant genodermatosis first described by brothers Hugh and William Hailey in 1939. Unlike autoimmune pemphigus vulgaris, HHD results from a genetic mutation disrupting keratinocyte adhesion, leading to chronic relapsing vesiculobullous eruptions primarily in intertriginous areas.
The condition typically manifests in the second to fourth decades, with complete penetrance but variable expressivity. It persists lifelong, exacerbated by friction, heat, sweat, and UV exposure, often causing significant morbidity due to pain, odour, and secondary infections.
Demographics
HHD affects all races and both sexes equally, with onset usually between ages 20-40, though paediatric and late-onset cases occur. Prevalence is estimated at 1 in 50,000, with sporadic cases from de novo mutations. Family history is present in most, following autosomal dominant inheritance with linkage to chromosome 3q21-24.
Clinical pathology
Lesions begin as flaccid vesicles and bullae in flexural sites—axillae, groin, inframammary folds, neck, and perianal region—rupturing to form erythematous, macerated plaques with crusted erosions and longitudinal rhagades (fissures). Central clearing yields annular or polycyclic patterns; chronic lesions thicken into vegetating plaques with malodour from bacterial overgrowth.
Nail dystrophy with white longitudinal bands and palmar pits may appear. Extracutaneous involvement is rare, but generalized erythroderma or fatal dissemination has been reported in severe cases. Triggers include sweating, friction, infection (Staphylococcus, Candida), and rarely drugs like vancomycin.
Histopathology
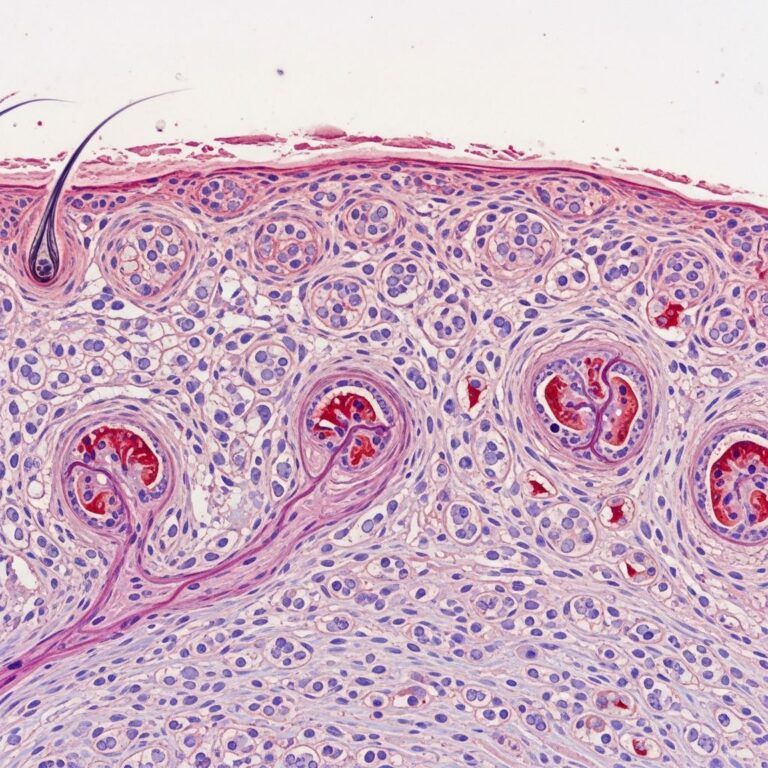
The hallmark is suprabasal acantholysis throughout the epidermis, creating a “dilapidated brick wall” or “row of tombstones” appearance—detached rounded keratinocytes with intact basal nuclei lining the floor of intraepidermal clefts.
Full-thickness acantholysis distinguishes HHD from pemphigus vulgaris (suprabasal only). Additional features include:
- Hyperkeratosis, parakeratosis, and spongiosis
- Dyskeratotic (corps ronds-like) cells
- Absent dermal-epidermal separation or eosinophils
- Negative direct immunofluorescence (DIF) for IgG/C3, ruling out autoimmunity
Electron microscopy reveals desmosomal dysfunction from defective calcium pumps.
Pathogenesis
HHD arises from mutations in ATP2C1 (SPTC1), encoding a Golgi P-type ATPase (SPCA1) that pumps Ca2+ into the Golgi for keratinocyte desmosome integrity. Over 100 mutations (missense, nonsense, deletions) impair Ca2+ homeostasis, reducing cell adhesion via E-cadherin/keratin 5/14 defects.
Variable expressivity stems from haploinsufficiency and environmental triggers disrupting the acantholysis threshold. Secondary infections amplify inflammation via cytokines (IL-1, IL-36).
Genetics
Autosomal dominant with near-complete penetrance; locus 3q22.1. Most cases inherit one mutated ATP2C1 allele, with loss-of-function leading to 50% pump activity. Genotyping confirms diagnosis in atypical cases.
Differential diagnosis
| Condition | Key Distinguishing Features |
|---|---|
| Pemphigus vulgaris | Autoimmune; oral involvement; positive DIF; suprabasal split only |
| Darier’s disease | Dyskeratosis > acantholysis; greasy keratotic papules; acrokeratosis |
| Intertrigo/impetigo | No family history; responds to hygiene/antibiotics; no acantholysis |
| Inverse psoriasis | Scales on non-intertriginous areas; Auspitz sign |
| Candida/tinea | KOH positive; satellite pustules |
Investigations
Diagnosis relies on clinical-family history and biopsy. DIF excludes pemphigus. PCR for ATP2C1 mutations aids atypical/genetic counselling cases. Cultures guide infection management.
Treatment pathology
No cure; management targets acantholysis, infection, and triggers:
- Topical: Superpotent steroids (clobetasol), calcipotriol, tacrolimus, benzoyl peroxide, bleach baths
- Systemic: Tetracyclines (doxycycline 100mg BD), naltrexone (low-dose 3-4.5mg), retinoids, immunosuppressants (methotrexate, cyclosporine)
- Procedural: Botulinum toxin for hyperhidrosis, CO2 laser, PDT, excision
Response correlates with reduced acantholysis on repeat biopsy.
Outcome
Chronic, non-scarring, but relapsing with flares. Morbidity from pain/odour impacts quality of life; rare squamous cell carcinoma or eczema herpeticum. Prognosis improves with trigger avoidance and multimodal therapy.
Frequently asked questions
What is the pathology of familial benign pemphigus?
Suprabasal full-thickness acantholysis with ‘tombstone’ keratinocytes, hyperkeratosis, and negative DIF.
Is Hailey-Hailey disease contagious?
No, it’s genetic, not infectious.
Can HHD be cured?
No, but symptoms are controllable with treatment.
What gene causes HHD?
ATP2C1 mutations on chromosome 3q.
Does HHD scar?
Typically no, lesions heal without atrophy.
References
- Benign Familial Pemphigus — JAMA Dermatology. 2022-01-19. https://jamanetwork.com/journals/jamadermatology/fullarticle/2787882
- Familial Benign Pemphigus — Patient.info. 2024. https://patient.info/doctor/dermatology/familial-benign-pemphigus
- Hailey-Hailey Disease (Benign Familial Pemphigus) — DermNet NZ. 2024-07-15. https://dermnetnz.org/topics/benign-familial-pemphigus
- Familial “Benign” Pemphigus? Erythroderma and Fatal Outcome — Anais Brasileiros de Dermatologia. 2019. https://www.anaisdedermatologia.org.br/en-familial-benign-pemphigus-erythroderma-fatal-articulo-S0365059619301497
- Benign Familial Pemphigus (Hailey-Hailey Disease) — StatPearls, NCBI Bookshelf. 2023. https://www.ncbi.nlm.nih.gov/books/NBK585136/
Similar Articles






Read full bio of medha deb