Germline BAP1 Mutation: 5 Cancer Risks, Diagnosis & Management
Understanding the hereditary BAP1 cancer syndrome: risks, skin tumours, and management strategies for affected individuals.

Author: Dr. Antonella Tosti, Reviewed by:
- Dr. Sarah Davies
- Dr. Papri Sarkar
What is the BAP1 gene?
BRCA1-associated protein-1 (**BAP1**) is a nuclear protein encoded by the BAP1 tumour-suppressor gene located on chromosome 3 (locus 3p21.1). The BAP1 protein functions as a deubiquitinating enzyme, which removes ubiquitin—a key regulator of protein degradation—from target proteins. This enzymatic activity is essential for numerous cellular processes, including:
- Transcriptional regulation
- Cell cycle control
- Differentiation
- DNA damage response
- Chromatin modification
BAP1 interacts with various protein complexes, such as the Polycomb repressive deubiquitinase complex and BRCA1, to maintain genomic stability and suppress tumour formation. Loss of BAP1 function disrupts these pathways, leading to uncontrolled cell proliferation and cancer predisposition.
What is germline BAP1 mutation?
Germline BAP1 mutations are inherited in an
autosomal dominant
pattern, requiring only one mutated allele from an affected parent for transmission to offspring. Each cell in the affected individual carries one mutant BAP1 allele alongside a wild-type (normal) allele. Full loss of BAP1 tumour-suppressor function occurs only after a ‘second hit’—a somatic mutation in the remaining wild-type allele—a process known as the two-hit hypothesis of tumourigenesis.These germline mutations define the
BAP1 cancer predisposition syndrome
(also called BAP1-inactivated tumour predisposition syndrome), first recognised in families with clusters of uveal melanoma and mesothelioma. Over 71 unique mutations spanning coding and non-coding regions of the BAP1 gene have been identified, often resulting in truncated or dysfunctional proteins.Who gets germline BAP1 mutation?
Germline BAP1 mutations are rare, with population prevalence estimates below 1 in 10,000. They occur worldwide without strong ethnic predisposition, though founder mutations have been reported in certain populations. Carriers are identified primarily through:
- Personal history of BAP1-associated cancers at young ages
- Family history of uveal melanoma, mesothelioma, or multiple melanocytic skin tumours
- Detection of multiple BAP1-inactivated melanocytic tumours (MBAITs) on skin examination
In reported cohorts of 215 patients from 87 families, malignancies manifested at younger median ages than in sporadic cases, underscoring the hereditary nature.
What are the cancer risks associated with germline BAP1 mutation?
The lifetime cancer risk in germline BAP1 carriers remains incompletely defined due to limited large-scale studies. However, aggregated data from 215 reported cases reveal the following malignancy spectrum:
| Cancer Type | Prevalence in Carriers | Notes |
|---|---|---|
| Uveal melanoma | 28% (60/215) | Most common; often aggressive with BAP1 loss |
| Mesothelioma | 22% (48/215) | Typically peritoneal; better prognosis than asbestos-related |
| Cutaneous melanoma | 18% (38/215) | May have shorter disease-free survival |
| Renal cell carcinoma | 9% (20/215) | Often aggressive clear cell subtype |
| Other (e.g., basal cell carcinoma, meningioma, breast, lung, pancreatic, thyroid) | <5% each | Less frequent associations |
Germline BAP1 mutations account for small fractions of these cancers overall: 1.6–4% of uveal melanomas, 1–2% of mesotheliomas, 0.01–0.63% of cutaneous melanomas, and 1.2% of renal cell carcinomas. Prognoses vary: worse outcomes in uveal melanoma and renal cell carcinoma; potentially better survival in mesothelioma.
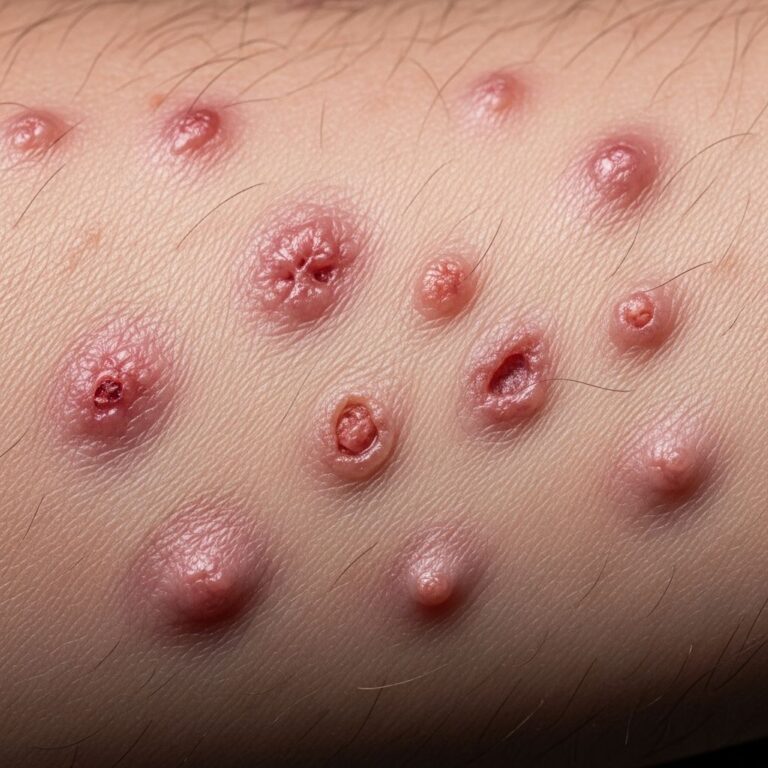
What is BAP1-inactivated melanocytic tumour?
The
BAP1-inactivated melanocytic tumour
(also termed melanocytic BAP1-mutated atypical intradermal tumour orMBAIT
) represents the most frequent and earliest clinical manifestation of germline BAP1 mutation, affecting 67–75% of carriers, usually after age 20. These benign but atypical naevi arise from biallelic BAP1 inactivation in melanocytes, combined frequently with BRAF mutations.Histologically, MBAITs feature small conventional naevus cells in the epidermis/dermis alongside larger epithelioid/spitzoid melanocytes lacking BAP1 expression on immunohistochemistry (IHC). Despite spitzoid features, they are molecularly distinct from atypical Spitz tumours.
Clinical features
MBAITs present as multiple (5–50), skin-coloured to tan-brown, dome-shaped papules (2–10 mm), often pedunculated, with a central pink structureless area on dermoscopy due to amelanotic epithelioid melanocytes. Common sites include trunk, limbs, and face; they arise rapidly over weeks.
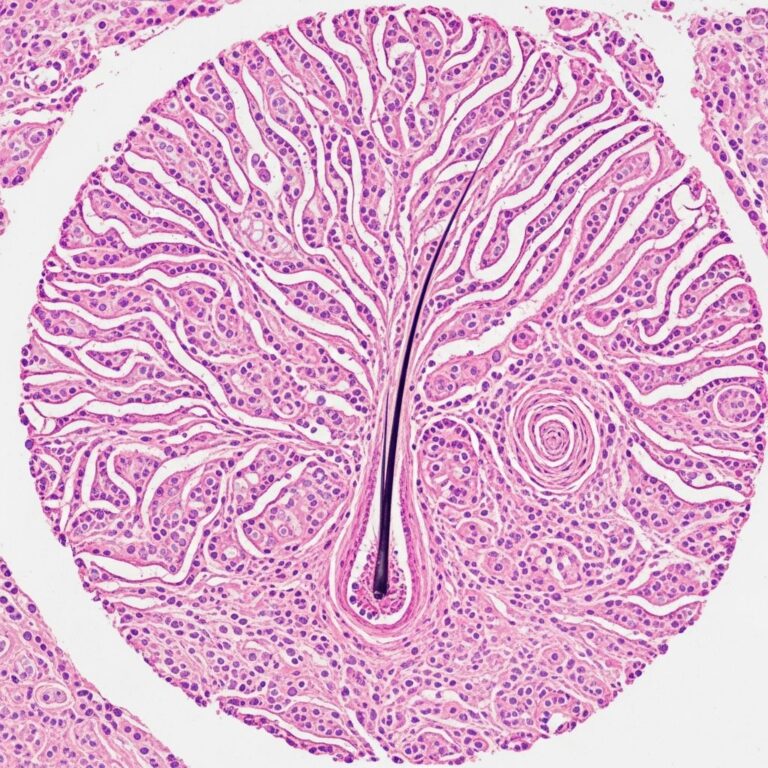
Histology of BAP1-inactivated melanocytic tumour
Key features include:
- Biphasic population: small type A naevus cells + large epithelioid type B melanocytes
- Biallelic BAP1 loss in type B cells (negative IHC staining)
- Frequent BRAF V600E mutation
- No high-risk melanoma features (e.g., deep mitoses, ulceration)
Despite multiplicity, progression to cutaneous melanoma is rare, as biallelic BAP1/ BRAF alterations alone are insufficient for frank malignancy.
Diagnosis of germline BAP1 mutation
Suspicion arises from:
- Multiple MBAITs (75% penetrance on total-body skin exam)
- Young-onset BAP1-associated cancers
- Family history of ≥2 relatives with syndrome cancers
Diagnostic steps:
- Total-body skin examination (TBSE) for MBAITs
- Excisional biopsy of suspicious lesions with BAP1 IHC
- Germline BAP1 sequencing (multigene panel or whole-exome)
- Consider tumour sequencing for somatic confirmation
Dermatologists play a pivotal role, as MBAITs precede malignancies by years.
Management of germline BAP1 mutation
Lesion management
Excision of symptomatic, changing, or cosmetically bothersome MBAITs. Routine removal of all lesions is unnecessary given low malignant potential.
Cancer surveillance
Evidence-based protocols (adapted from expert consensus):
| Organ/Screening | Frequency | Age to Start |
|---|---|---|
| Skin: TBSE + dermoscopy | 6–12 months | Birth |
| Eye: Annual dilated exam | Yearly | 11 years |
| Abdomen: MRI or US | Every 2 years | 30–35 years |
| Chest: Low-dose CT | Every 2 years | 30–35 years (if asbestos exposure) |
Genetic counselling for carriers and at-risk relatives is essential.
Frequently asked questions
What causes BAP1-inactivated melanocytic tumours?
Germline BAP1 mutation plus somatic second hit in melanocytes, often with BRAF co-mutation.
Are BAP1-inactivated melanocytic tumours cancerous?
No, they are benign atypical naevi despite unusual histology; malignant transformation is exceedingly rare.
Should all MBAITs be removed?
Only if symptomatic, changing, or diagnostic uncertainty; surveillance preferred for stable lesions.
What is the prognosis for BAP1 carriers?
High lifetime cancer risk but variable by tumour type; early detection improves outcomes.
Is genetic testing recommended?
Yes, for those with multiple MBAITs, young-onset syndrome cancers, or suggestive family history.
References
- Genotypic and Phenotypic Features of BAP1 Cancer Syndrome — JAMA Dermatology. 2019-04-17. https://jamanetwork.com/journals/jamadermatology/fullarticle/2648082
- Germline BAP1 mutation and BAP1 inactivated melanocytic tumours — DermNet NZ. 2023-01-01. https://dermnetnz.org/topics/germline-bap1-mutation
- BAP1-TPDS: BAP1 Tumor Predisposition Syndrome — National Cancer Institute (NCI.gov). 2024-06-15. https://www.ncbi.nlm.nih.gov/books/NBK547752/
- Hereditary BAP1-associated tumor predisposition syndrome — Genetics Home Reference (NIH.gov). 2023-11-20. https://medlineplus.gov/genetics/condition/hereditary-bap1-associated-tumor-predisposition-syndrome/
- BAP1 tumor predisposition syndrome — American Society of Clinical Oncology (ASCO). 2024-02-10. https://www.cancer.net/cancer-types/bap1-tumor-predisposition-syndrome
Similar Articles






Read full bio of Sneha Tete