IgG4-Related Skin Disease: 3 Key Diagnostic Criteria
Comprehensive pathology guide to IgG4-related skin disease: histological features, diagnostic criteria, and clinical insights.

IgG4-Related Skin Disease Pathology
IgG4-related skin disease represents a rare manifestation of immunoglobulin G4-related disease (IgG4-RD), a systemic fibroinflammatory condition characterized by dense lymphoplasmacytic infiltrates rich in IgG4-positive plasma cells, storiform fibrosis, and often obliterative phlebitis. While IgG4-RD commonly affects organs like the pancreas, salivary glands, and orbits, cutaneous involvement is infrequent, occurring in less than 10% of cases, and typically presents alongside systemic features.
Introduction to IgG4-Related Disease
IgG4-RD is an immune-mediated disorder first comprehensively described in the early 2000s, initially recognized in the context of autoimmune pancreatitis and sclerosing sialadenitis. It is marked by tumefactive lesions—swellings or pseudotumors—that mimic malignancy or infection. Pathologically, the hallmark triad includes: (1) dense infiltration of IgG4-positive plasma cells, (2) storiform (whorled or cartwheel-like) fibrosis, and (3) obliterative phlebitis (occlusion of small veins by inflammatory cells and fibrosis). Eosinophils are frequently abundant, and serum IgG4 levels are elevated in up to 70% of patients.
In the skin, these features translate to a spectrum of lesions, from erythematous papules and plaques on the head and neck to nodular indurations and purpuric eruptions. Cutaneous IgG4-RD is classified into primary (direct plasma cell infiltration forming mass lesions) and secondary (IgG4-mediated inflammation without prominent plasma cell masses) types, aiding in precise histopathological diagnosis.
Clinical Features
Cutaneous manifestations of IgG4-RD are rare and often signal multisystem involvement. A systematic review indicates over 60% of reported cases originate from Japan, predominantly affecting males in their sixth decade. Lesions typically appear as pruritic erythematous papules, plaques, or nodules, favoring the head and neck region (periauricular, cheek, mandible). Other presentations include purpura, patches, prurigo nodularis-like nodules, psoriasis-like eruptions, hypergammaglobulinemic purpura on lower extremities, urticarial vasculitis, and rarely ischemic digits (Raynaud phenomenon or gangrene).
Systemic associations are common, particularly head-and-neck involvement: submandibular/parotid sialadenitis, lacrimal/orbital dacryoadenitis (Mikulicz disease with palpebral swelling, sicca syndrome, exophthalmos). Skin lesions may precede systemic disease by months to years, emphasizing the need for vigilance in dermatologic practice.
- Primary cutaneous subtypes: Cutaneous plasmacytosis (trunk/limbs papulonodules), pseudolymphoma/angiolymphoid hyperplasia with eosinophilia (ALHE; periauricular plaques), Mikulicz disease.
- Secondary subtypes: Psoriasis-like, maculopapular eruptions, purpura, urticarial vasculitis, ischemic digits.
Pruritus is reported in most cases, with rapid steroid responsiveness but frequent relapse on tapering.
Histopathology
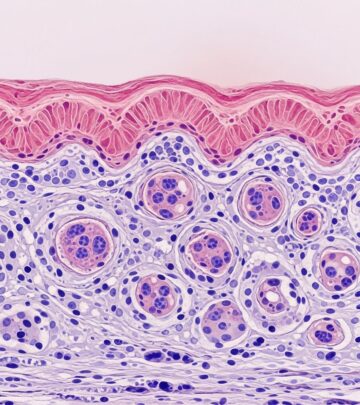
Histological examination is pivotal for diagnosis. Skin biopsies reveal a dense lymphoplasmacytic infiltrate in the dermis, often extending to subcutaneous tissue. Infiltrates are perivascular, periadnexal, and nodular, with numerous plasma cells (IgG4+/IgG+ ratio >40%, >10-50 IgG4+ cells/high-power field [HPF]). Germinal centers and lymphoid follicles may form, especially in head/neck biopsies.
The classic storiform fibrosis—irregular, storiform (mat-like) collagen bundles—is less common in skin than visceral organs but highly specific when present. Obliterative phlebitis, with vein lumina narrowed by fibrous tissue and inflammatory cells, is rare in cutaneous samples (0-1% in mimics). Eosinophils are prominent, and immunohistochemistry (IHC) confirms IgG4 expression: ideally >200 IgG4+ cells/HPF in primaries, though skin thresholds are lower (10-50/HPF).
| Feature | Description | Skin Prevalence |
|---|---|---|
| Lymphoplasmacytic Infiltrate | Dense dermal/subcutaneous plasma cells, IgG4+ >40% of IgG+ | Common (high in mimics) |
| Storiform Fibrosis | Whorled, cartwheel collagen pattern | Rare (specificity high) |
| Obliterative Phlebitis | Vein occlusion by inflammation/fibrosis | Very rare (0% in many studies) |
| Eosinophils | Increased tissue eosinophils | Frequent |
| IHC Threshold | >10-50 IgG4+ cells/HPF; ratio >0.4 | Required for diagnosis |
Primary lesions show massive direct plasma cell infiltration fulfilling comprehensive criteria (>40% IgG4+/IgG+, >10/HPF). Secondary lesions exhibit perivascular IgG4 deposition without mass formation.
Classification of IgG4-Related Skin Disease
Yokura et al. proposed a practical classification dividing lesions into primary (mass-forming, direct infiltration) and secondary (indirect IgG4 effects). This framework guides diagnosis:
Primary Lesions
- Cutaneous plasmacytosis: Multiple red-brown papulonodules/indurations on trunk/proximal limbs.
- Pseudolymphoma/ALHE: Plaques/nodules on periauricular/cheek/mandible areas, with eosinophils/vascular proliferation.
- Mikulicz disease: Orbital/lacrimal swelling with sicca/exophthalmos.
Secondary Lesions
- Psoriasis-like eruptions (scaly plaques mimicking vulgaris).
- Unspecified maculopapular/erythematous rashes.
- Hypergammaglobulinemic purpura (asymmetric lower leg purpura).
- Urticarial vasculitis (prolonged wheals ± purpura).
- Ischemic digits (Raynaud/gangrene, rare).
This distinction is crucial: primaries meet strict IHC criteria; secondaries show milder infiltrates.
Diagnostic Criteria
No universal skin-specific criteria exist, but adapted comprehensive guidelines require:
- Characteristic histological findings (lymphoplasmacytic infiltrate ± storiform fibrosis/phlebitis).
- IgG4+ plasma cells >10/HPF (skin minimum; >30-50/HPF preferred), IgG4+/IgG+ >40% (averaged over 3 HPFs).
- Exclusion of mimics via clinicopathologic correlation.
Serum IgG4 elevation supports but is not diagnostic (elevated in 70%, normal in 30%). Biopsy from representative lesions is essential.
Differential Diagnosis
The broad differential reflects overlapping lymphoplasmacytic histology:
- Neoplastic: Primary cutaneous marginal zone lymphoma (PCMZL), Rosai-Dorfman disease, low-grade B-cell lymphomas.
- Inflammatory: Cutaneous plasmacytosis, Sjögren syndrome, ALHE, granuloma faciale, erythema elevatum diutinum (EED), sarcoidosis, pseudolymphomas, syphilis, lichen sclerosus, necrobiosis lipoidica, psoriasis, erosive pustular dermatosis.
- Other: Histiocytosis, lymphoplasmacytic plaque.
Lymphoplasmacytic infiltrates are nonspecific (common in 69 mimics), but storiform fibrosis/phlebitis and strict IHC distinguish IgG4-RD. No mimics met >200 IgG4+ cells/HPF; only 13-23% met ratio/>10/HPF thresholds.
| Entity | Key Overlap | Distinguishing Features |
|---|---|---|
| ALHE | Eosinophils, vascularity | Lacks storiform fibrosis; epithelioid endothelial cells |
| PCMZL | Plasma cells, follicles | Light chain restriction, clonality |
| EED | Fibrosis, phlebitis | Neutrophils, leukocytoclastic; rare storiform |
| Plasmacytosis | Plasma cell nodules | Polyclonal; less IgG4-dominant |
Treatment and Prognosis
First-line therapy is systemic corticosteroids (e.g., prednisone 0.6-1 mg/kg), yielding rapid lesion resolution (>80% response). Relapses occur on taper (50-70%), necessitating steroid-sparing agents: rituximab (anti-CD20, excellent for relapses), azathioprine, methotrexate. Topical/intralesional steroids suffice for localized disease. Monitoring includes serum IgG4, imaging for systemic sites.
Prognosis is favorable with treatment, though fibrosis may persist. Early recognition prevents organ damage.
Frequently Asked Questions (FAQs)
Is cutaneous IgG4-RD common?
No, it affects <10% of IgG4-RD patients and is often systemic.
What is the minimum IgG4+ cell count for skin diagnosis?
>10/HPF with IgG4+/IgG+ >40%; higher thresholds preferred.
Does serum IgG4 confirm diagnosis?
No, elevated in 70%; normal levels occur. Histology is key.
Are skin lesions steroid-responsive?
Yes, rapidly, but relapses are common on tapering.
How to differentiate from lymphoma?
IHC (polyclonality, no light chain restriction), fibrosis/phlebitis.
References
- IgG4-related skin diseases: A brief review — Journal of Surgical and Specialty Disciplines. 2023. https://jsstd.org/igg4-related-skin-diseases-a-brief-review/
- Assessment of specificity of dermatopathologic criteria for IgG4-related skin disease — Journal of Cutaneous Pathology (Wiley). 2023-10-18. https://onlinelibrary.wiley.com/doi/10.1111/cup.14548
- Cutaneous and systemic IgG4-related disease: a review — PubMed / Journal of Cutaneous Pathology. 2019-07-22. https://pubmed.ncbi.nlm.nih.gov/31329383/
- IgG4-related disease: A mini-review — Rare Diseases Journal. 2022. https://www.rarediseasesjournal.com/articles/igg4-related-disease-a-minireview.pdf
- IgG4-related skin disease pathology — DermNet NZ. 2023. https://dermnetnz.org/topics/igg4-related-skin-disease-pathology
- IgG4-related disease — DermNet NZ. 2023. https://dermnetnz.org/topics/igg4-related-disease
Similar Articles

Read full bio of Sneha Tete



