Juvenile Systemic Granulomatosis: Blau Syndrome
Understanding juvenile systemic granulomatosis and Blau syndrome: genetics, symptoms, and diagnosis.

Juvenile Systemic Granulomatosis and Blau Syndrome
Juvenile systemic granulomatosis, commonly referred to as Blau syndrome, is a rare monogenic autoinflammatory disorder that primarily affects the skin, joints, and eyes in children. This inherited condition results from mutations in the CARD15 gene (also known as NOD2) and is characterized by the formation of non-caseating granulomas in multiple organ systems. The disease typically manifests early in childhood, usually before age 4, and affects both males and females equally.
Genetic Basis and Inheritance
Juvenile systemic granulomatosis results from mutations in the NACHT domain of the CARD15 (caspase recruitment domain 15) gene, also known as the NOD2 gene, located on chromosome 16q12.1-13. To date, identified mutations have consistently involved heterozygous single-nucleotide substitutions. The NOD2 protein plays a crucial role in immune regulation, and when mutated, it becomes overactive and alters how the immune system functions. This overreaction triggers an extreme inflammatory response that cascades through multiple body systems.
Blau syndrome follows an autosomal dominant inheritance pattern, meaning that a child who has one parent with the condition or who carries the gene mutation has a 50% chance of inheriting the altered gene and developing the syndrome. Importantly, a child only needs to inherit one mutated copy of the gene to develop the disease, as the mutation is dominant.
Clinical Manifestations
Juvenile systemic granulomatosis presents with a characteristic triad of clinical features affecting three primary organ systems: skin, joints, and eyes. The specific manifestations and their severity can vary significantly from person to person, and symptoms may change as the disease progresses.
Skin Manifestations
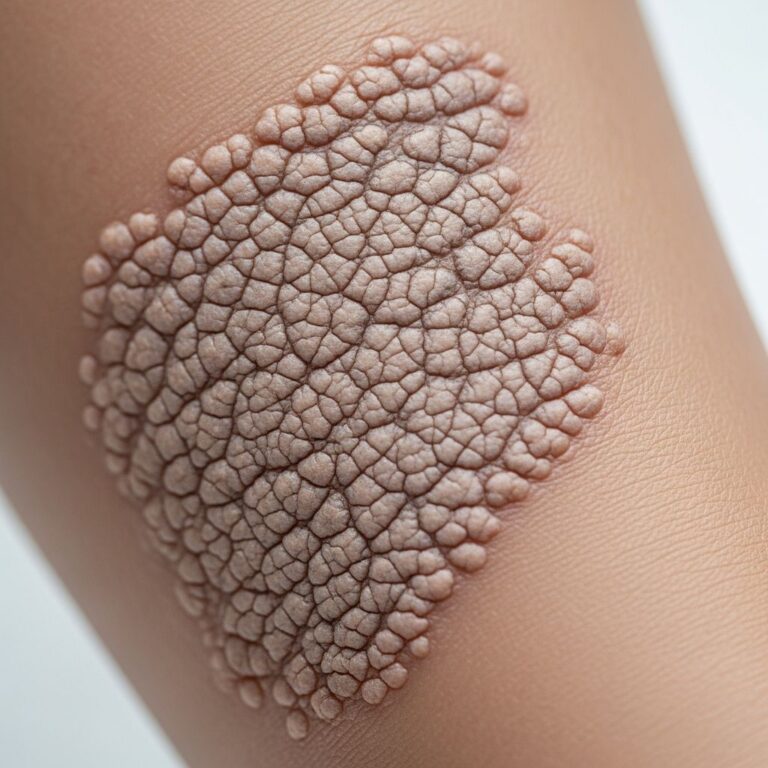
Granulomatous dermatitis is typically the earliest clinical sign of Blau syndrome, often appearing as the initial presentation. This form of skin inflammation presents as a persistent rash that can be scaly or involve hard lumps (nodules) that can be felt under the skin. The rash is usually found on the torso, arms, and legs. Skin biopsy reveals noncaseating granulomatous dermatitis, showing a particular pattern of mixed inflammatory infiltrate characteristic of the disease. The granulomas are found in the upper dermis and are often located around hair follicles.
The skin lesions in juvenile systemic granulomatosis are distinguished by their granulomatous nature rather than caseating necrosis, which helps differentiate this condition from other granulomatous diseases such as tuberculosis.
Joint Manifestations
Joint involvement is the most common feature, occurring in approximately 90% of patients. The arthritis typically presents as symmetrical polyarthritis with the following characteristics:
- Boggy, nontender joint swelling
- Wrists, knees, and ankles most commonly affected
- Large and small joints may be involved
- Inflammation of tendons around the joints
- Fluid accumulation around affected joints
- Potential for permanent bending of toes and fingers in advanced cases
Non-caseating granulomas are identified in biopsies from the synovium of affected joints, confirming the granulomatous nature of the arthritis.
Ocular Manifestations
Eye involvement is a significant feature of Blau syndrome, with inflammatory processes affecting ocular structures. Common ophthalmic manifestations include:
- Inflammation of the eye’s middle layer (uvea), called uveitis
- Conjunctivitis (sometimes called pink eye)
- Eye pain and redness
- Decreased vision or visual field abnormalities
- Diplopia (double vision)
Non-caseating granulomas are also observed in eye biopsies and other involved ocular tissues, consistent with the systemic granulomatous nature of the condition.
Associated Systemic Manifestations
Beyond the classic triad of skin, joint, and eye involvement, juvenile systemic granulomatosis can affect multiple other organ systems, particularly in severe or progressive cases:
- Cardiovascular: Increased pressure in the blood vessels of the heart and lungs
- Renal: Kidney disease and calcium deposits in the kidneys
- Systemic symptoms: Fevers, general malaise, and overall feeling of ill health
- Neurological: Nerve damage and peripheral neuropathy
- Other complications: Arthritis and inflammation affecting various connective tissues
Pathological Features
The hallmark histopathological finding in juvenile systemic granulomatosis is the presence of non-caseating granulomas throughout affected tissues. These granulomas are collections of activated immune cells that form in response to the pathogenic immune activation caused by the NOD2 mutation.
Skin biopsy demonstrates noncaseating granulomatous dermatitis with a mixed inflammatory infiltrate pattern. The granulomas are predominantly found in the upper dermis and frequently surround hair follicles. This pattern is characteristic and helps establish the diagnosis when combined with clinical presentation and genetic testing.
The absence of caseating necrosis—the central tissue death seen in conditions like tuberculosis—is an important distinguishing feature that aids in differential diagnosis.
Diagnostic Approach
Diagnosis of juvenile systemic granulomatosis involves a combination of clinical assessment, histopathological examination, and genetic testing. Key diagnostic components include:
- Clinical evaluation: Assessment of the characteristic triad of skin rash, arthritis, and eye inflammation, typically beginning before age 4
- Skin biopsy: Demonstrates non-caseating granulomatous dermatitis with characteristic upper dermal granulomas
- Ocular examination: Evaluation of uveal inflammation and conjunctival involvement
- Joint assessment: Clinical examination and imaging of affected joints to confirm polyarthritis
- Genetic testing: CARD15/NOD2 gene sequencing to identify the causative mutation
- Systemic evaluation: Assessment of other potential organ involvement including renal and cardiovascular systems
Differential Diagnosis
Juvenile systemic granulomatosis must be differentiated from several other conditions with overlapping features, including granulomatosis with polyangiitis (GPA), sarcoidosis, and other autoinflammatory syndromes. The presence of non-caseating granulomas helps distinguish this condition from infectious granulomatous diseases. The specific genetic mutation in the CARD15 gene and the characteristic clinical triad in young children help confirm the diagnosis.
Disease Progression and Complications
The clinical course of juvenile systemic granulomatosis can be highly variable. Some patients experience mild disease limited to skin and joint manifestations, while others develop progressive systemic involvement. Potential long-term complications may include:
- Chronic arthritis with joint damage and deformity
- Visual impairment from chronic ocular inflammation
- Kidney involvement and potential renal dysfunction
- Cardiovascular complications from vascular inflammation
- Permanent bending or contractures of fingers and toes
Current Management Considerations
While specific treatment protocols continue to evolve, management of juvenile systemic granulomatosis typically focuses on controlling inflammation and preventing long-term complications. Immunosuppressive therapies are generally employed to modulate the overactive immune response characteristic of this condition. The overactivity of the NOD2 protein is central to disease pathogenesis, making immune regulation a key therapeutic target.
Early diagnosis and intervention are important to prevent organ damage and manage symptoms effectively. Treatment decisions should be individualized based on disease severity and organ involvement, with close monitoring for disease progression and treatment response.
Frequently Asked Questions
Q: At what age does juvenile systemic granulomatosis typically appear?
A: Blau syndrome typically manifests in childhood, usually before age 4, though the exact timing can vary among affected individuals.
Q: Is juvenile systemic granulomatosis inherited?
A: Yes, Blau syndrome is an autosomal dominant genetic condition. If one parent carries the CARD15 mutation, there is a 50% chance their child will inherit the gene and develop the condition. Only one mutated copy is needed to cause the disease.
Q: What are the three primary features of Blau syndrome?
A: The characteristic triad includes granulomatous skin rash (usually on torso, arms, and legs), symmetrical polyarthritis (especially affecting wrists, knees, and ankles), and ocular inflammation (uveal inflammation and conjunctivitis).
Q: How is juvenile systemic granulomatosis diagnosed?
A: Diagnosis is established through clinical evaluation of the characteristic triad, skin biopsy showing non-caseating granulomas, and genetic testing for CARD15/NOD2 mutations. Systemic assessment and ophthalmologic evaluation are also important components.
Q: Can juvenile systemic granulomatosis affect organs other than skin, joints, and eyes?
A: Yes, while skin, joints, and eyes are the primary organs affected, Blau syndrome can involve other systems including the kidneys, cardiovascular system, and nervous system in some patients, particularly in severe cases.
Q: What distinguishes juvenile systemic granulomatosis from other granulomatous diseases?
A: The key distinguishing feature is the presence of non-caseating granulomas (without central necrosis), combined with the specific CARD15 gene mutation, the autosomal dominant inheritance pattern, and the characteristic early childhood onset with the triad of skin, joint, and eye involvement.
References
- Juvenile Systemic Granulomatosis (Blau Syndrome) — DermNet NZ. 2024. https://dermnetnz.org/topics/juvenile-systemic-granulomatosis
- Blau Syndrome — MedlinePlus Genetics, National Library of Medicine. 2024. https://medlineplus.gov/genetics/condition/blau-syndrome/
- Blau Syndrome: Definition, Symptoms, and Treatment — Medical News Today. 2023. https://www.medicalnewstoday.com/articles/blau-syndrome
- Blau Syndrome: Symptoms, Causes & Outlook — Cleveland Clinic. 2025. https://my.clevelandclinic.org/health/diseases/25182-blau-syndrome
- Blau Syndrome (Juvenile Systemic Granulomatosis): State-of-the-Art Review — NIH PubMed Central. 2024. https://pmc.ncbi.nlm.nih.gov/articles/PMC12285560/
Similar Articles






Read full bio of Sneha Tete