Kindler Syndrome: Causes, Symptoms, and Management
Understanding Kindler syndrome: A rare hereditary blistering disorder with progressive skin changes.

What is Kindler Syndrome?
Kindler syndrome (KS) is a rare form of epidermolysis bullosa characterized by a mixed pattern of blistering on multiple levels within and beneath the basement membrane zone. This hereditary condition affects skin integrity and fragility, presenting with distinctive clinical features that evolve throughout the patient’s lifetime. Kindler syndrome represents the fourth major type of epidermolysis bullosa, alongside simplex, junctional, and dystrophic forms.
The disorder is caused by mutations in the FERMT1 gene, which encodes the protein kindlin-1. This protein plays a crucial role in binding actin to the extracellular matrix, and its loss of function leads to the characteristic skin manifestations observed in affected individuals. Understanding the genetic basis of Kindler syndrome has opened new diagnostic and therapeutic possibilities for managing this challenging condition.
Genetics and Inheritance Pattern
Kindler syndrome is inherited as an autosomal recessive disorder. This inheritance pattern means that an affected individual must inherit one mutated gene from each parent. Parents are typically carriers of the gene mutation but do not express the disease phenotype. On average, one in four children born to two carrier parents will be affected by the condition, while two will be carriers and one will inherit two normal gene copies.
The genetic defect has been identified on the short arm of chromosome 20, specifically in the FERMT1 gene. Since the discovery of the genetic basis of Kindler syndrome, researchers have identified numerous mutations within this gene. The condition is quite rare, with more than 120 cases reported in medical literature since its original description by Kindler in 1954 in a 14-year-old girl who presented with unusual blistering on the hands, arms, feet, and legs followed by photosensitivity and pigmentary changes.
Clinical Features and Manifestations
The clinical presentation of Kindler syndrome typically begins in infancy and progresses through distinct phases. Early manifestations differ significantly from later-stage features, reflecting the dynamic nature of this genodermatosis.
Early Childhood Features
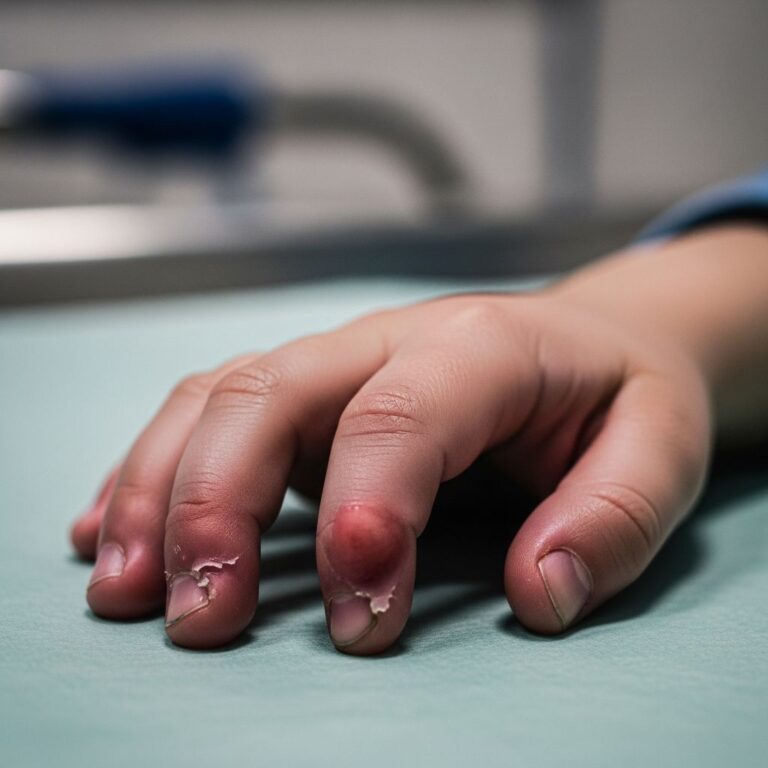
Kindler syndrome classically presents with acral blistering in infancy, particularly on the backs of the hands and tops of the feet. These blisters are typically induced by trauma or minor friction and occur predominantly in the first months of life. Affected infants often experience difficulty with routine activities such as feeding, which can be complicated by oral blistering and mucosal involvement.
The blisters tend to improve with age, though repeated trauma can cause complications. Scarring from recurrent blistering on the hands may result in fusion of skin between the fingers and toes, a condition known as pseudosyndactylyl. Additionally, affected children may develop nail dystrophy, characterized by abnormal nail growth and appearance.
Progressive Features
As children with Kindler syndrome age, the condition evolves with the development of progressive skin changes. Key features include:
- Poikiloderma: Progressive poikiloderma develops, especially on sun-exposed areas, with a characteristic pattern of atrophy, dyspigmentation, and telangiectasia.
- Skin atrophy: Thin, papery skin develops starting on the hands and feet and later affecting other body areas. The skin develops a distinctive “cigarette paper-like” appearance with fine wrinkling.
- Photosensitivity: Photosensitivity is prominent in childhood but improves with age. However, affected individuals remain predisposed to malignant skin tumors in adulthood due to cumulative sun exposure and genetic predisposition.
- Hyperkeratosis: Thickening of the skin on palms and soles, particularly over pressure areas like heels.
- Telangiectasia: Small clusters of visible blood vessels just under the skin develop, giving the skin a mottled appearance.
Mucosal and Systemic Manifestations
Beyond cutaneous features, Kindler syndrome frequently involves mucosal surfaces and other body systems. Mucosal involvement usually begins in adolescence, with the oral mucosa most commonly affected. Clinical manifestations include:
- Hemorrhagic gingivitis and periodontal disease
- Poor dentition and premature tooth loss due to gingival fragility
- Chronic constipation
- Esophageal, urethral, and anal strictures
- Severe colitis
- Conjunctivitis and ectropion
- Phimosis (in affected males)
Diagnostic Approach
Early clinical diagnosis of Kindler syndrome can be challenging, as many early clinical and pathological features closely resemble other types of epidermolysis bullosa, particularly epidermolysis bullosa simplex. A comprehensive diagnostic approach is essential for accurate classification and appropriate management.
Histopathological Findings
Light microscopic examination reveals characteristic features that help distinguish Kindler syndrome from other blistering disorders:
- Epidermal atrophy
- Vacuolar alteration with cleft formation
- Colloid bodies
- Dilated capillaries in the superficial dermis
- Dermal edema or mild fibrosis
- Clumping of tonofilaments surrounding the nucleus
Notably, hemidesmosomes, desmosomes, and anchoring fibrils are normal, distinguishing KS from other epidermolysis bullosa forms.
Electron Microscopy
Transmission electron microscopy reveals characteristic ultrastructural features of Kindler syndrome, including reduplication of the basal lamina with interspersed zones of collagen deposition. At sites of disruption, fibroblast-like cells are observed lying within clefts beneath exposed keratinocytes, a distinctive finding not seen in other EB types.
Immunofluorescence Microscopy
Immunofluorescence microscopy studies using carboxy-terminal anti-kindlin-1 antibodies serve as a valuable diagnostic tool. Studies have demonstrated that marked reduction or complete absence of immunostaining is consistently observed in patients with mutations in the KIND1 gene. This anti-kindlin-1 antibody approach has proven to be a reliable and consistent marker for diagnosing Kindler syndrome, helping to differentiate it from other epidermolysis bullosa variants.
Differential Diagnosis
Several conditions must be distinguished from Kindler syndrome, as they may present with overlapping features:
- Bloom syndrome: Characterized by growth deficiency and immunodeficiency, not typically seen in KS
- Cockayne syndrome: Features neurological abnormalities absent in KS
- Dyskeratosis congenita: Presents with the classic triad of reticulated hyperpigmentation, nail dystrophy, and leukoplakia
- Epidermolysis bullosa simplex: Shares early blistering but lacks progressive poikiloderma and photosensitivity
- Rothmund-Thomson syndrome: Features poikiloderma but lacks significant blistering in infancy
- Xeroderma pigmentosum: Although photosensitivity and poikilodermatous changes overlap, acral bullae are not seen, and patients experience early-onset skin cancers with neurological abnormalities
Molecular Pathogenesis
The underlying molecular defect in Kindler syndrome represents a unique mechanism distinct from other epidermolysis bullosa types. While epidermolysis bullosa simplex, junctional, and dystrophic forms result from defects in the keratin-intermediate filament-hemidesmosome complex network at the basement membrane, Kindler syndrome is the first genodermatosis caused by a defect in the structural link between the actin cytoskeleton, focal contacts, and extracellular matrix.
The loss of kindlin-1 function disrupts the critical connection between intracellular actin filaments and the extracellular matrix. This actin-extracellular matrix disruption initially produces a phenotypic mimic of keratin-extracellular matrix mutations seen in other epidermolysis bullosa forms, explaining the diagnostic confusion in early disease presentation. However, the distinct ultrastructural findings and progression pattern help differentiate Kindler syndrome from other EB variants.
Disease Progression and Prognosis
Kindler syndrome demonstrates a characteristic pattern of disease evolution across the patient’s lifetime. While blistering and photosensitivity tend to diminish in severity with age, poikiloderma is permanent. Scarring is usually minimal; however, repeated trauma can result in development of scar tissue, particularly affecting the hands and feet.
A critical concern in adult patients with Kindler syndrome is the increased predisposition to malignant skin tumors. Cumulative sun exposure throughout life, combined with the underlying genetic defect, significantly elevates the risk of developing squamous cell carcinoma and other skin cancers. This necessitates vigilant dermatologic surveillance and aggressive photoprotection strategies throughout the patient’s lifetime.
Management and Treatment Strategies
Currently, there is no cure for Kindler syndrome. Management focuses on symptom relief, prevention of complications, and improvement of quality of life through a multidisciplinary approach.
Photoprotection
Given the photosensitivity component of Kindler syndrome, comprehensive photoprotection is essential. Strategies include:
- Daily use of broad-spectrum sunscreen with SPF 30 or higher
- Protective clothing including long sleeves, pants, and wide-brimmed hats
- Avoidance of peak sun exposure hours
- Use of protective eyewear to prevent conjunctival involvement
Skin Care
Gentle skin care is paramount for managing skin fragility and preventing blistering. This includes avoiding friction, maintaining skin hydration with emollients, and using soft, non-irritating clothing. Trauma avoidance is critical, as minor injuries can trigger blister formation.
Oral and Dental Management
Regular dental evaluation and aggressive preventive dentistry are essential components of care. Early intervention for periodontal disease, appropriate oral hygiene techniques, and consideration of dental prosthetics may be necessary as the condition progresses.
Mucosal Involvement
Management of esophageal strictures, urethral stenosis, and other mucosal complications may require endoscopic intervention or surgical correction. Nutritional support and assessment for malabsorption may be necessary in patients with severe gastrointestinal involvement.
Frequently Asked Questions
Q: Is Kindler syndrome fatal?
A: While Kindler syndrome significantly impacts quality of life, it is not directly fatal. However, complications such as malignant transformation and severe systemic involvement require careful management to prevent life-threatening scenarios.
Q: Can Kindler syndrome be cured?
A: Currently, there is no cure for Kindler syndrome. Treatment is focused on managing symptoms, preventing complications, and improving quality of life through comprehensive multidisciplinary care.
Q: What is the life expectancy for someone with Kindler syndrome?
A: Life expectancy varies depending on disease severity and management of complications. With appropriate care and surveillance, many individuals with Kindler syndrome have normal or near-normal lifespans.
Q: How is Kindler syndrome inherited?
A: Kindler syndrome is inherited in an autosomal recessive pattern, meaning an affected individual must inherit one mutated gene from each parent. Parents are typically unaffected carriers.
Q: What genetic testing is available for Kindler syndrome?
A: Genetic testing for FERMT1 mutations is available and can confirm the diagnosis of Kindler syndrome. Genetic counseling is recommended for affected families to discuss inheritance patterns and recurrence risks.
References
- Kindler epidermolysis bullosa: MedlinePlus Genetics — U.S. National Library of Medicine. 2024. https://medlineplus.gov/genetics/condition/kindler-epidermolysis-bullosa/
- Kindler Syndrome: A New Mutation and New Diagnostic Possibilities — JAMA Dermatology. 2005. https://jamanetwork.com/journals/jamadermatology/fullarticle/404917
- Kindler Syndrome: A Multidisciplinary Management Approach — Actas Dermo-Sifiliográficas. 2020. http://www.actasdermo.org/en-kindler-syndrome-a-multidisciplinary-management-articulo-S157821902030281X
- Kindler syndrome — Indian Journal of Dermatology, Venereology and Leprology. 2018. https://ijdvl.com/kindler-syndrome/
- Clinical insights and therapeutic strategies in Kindler syndrome — International Journal of Rare Disorders. 2023. https://www.ijord.com/index.php/ijord/article/view/1854
Similar Articles






Read full bio of medha deb