Malignant Atrophic Papulosis: Diagnosis, Treatment, Prognosis
Rare vasculopathy with characteristic skin lesions that may progress to life-threatening systemic involvement.

Malignant atrophic papulosis (MAP), also known as Degos disease or Kohlmeier-Degos disease, is a rare and potentially lethal thrombo-occlusive vasculopathy primarily affecting small and medium-sized arteries of the skin, gastrointestinal tract, central nervous system, and other organs. Characterized by pathognomonic skin lesions—small papules with porcelain-white atrophic centers surrounded by erythematous, telangiectatic rims—MAP exists in benign (cutaneous-limited) and malignant (systemic) forms. The benign form remains confined to the skin in about 15% of cases, while the systemic variant can lead to fatal complications such as intestinal perforation, cerebrovascular events, or pericardial involvement. First described in the 1940s by Robert Degos, the disease has an estimated incidence of less than 1 in 100,000, with a male predominance and typical onset between ages 20 and 50. Early recognition of skin lesions is crucial, as systemic involvement can emerge months to years later, often within the first 7 years.
What is Malignant Atrophic Papulosis?
MAP is an uncommon vasculopathy involving endothelial dysfunction, coagulopathy, and thrombosis, leading to characteristic infarcts in affected tissues. It manifests with cutaneous lesions that are nearly pathognomonic, but similar pathology can occur internally, causing ischemia and necrosis. The disease is stratified into benign atrophic papulosis (BAP), limited to skin, and malignant AP (MAP), with visceral involvement. Systemic MAP affects small- and medium-sized arteries, with thrombotic occlusions visible histologically. Cutaneous-only cases have been reported, but up to one-third of patients develop systemic symptoms. Lesions average around 30 per patient but can exceed 600. Transformation from cutaneous to systemic forms can occur rapidly, emphasizing vigilant monitoring.
Who Gets Malignant Atrophic Papulosis (Epidemiology)?
MAP is exceedingly rare, with fewer than 300 cases documented worldwide. It predominantly affects young to middle-aged adults (20-50 years), with a 2:1 male-to-female ratio. No strong ethnic predisposition exists, though cases are reported globally. Familial clusters are rare, suggesting primarily sporadic occurrence, though genetic factors like mutations in TREX1 or associations with connective tissue diseases (e.g., systemic lupus erythematosus, scleroderma) have been noted. Risk factors remain poorly defined, but coagulopathies and autoimmune overlaps increase suspicion. Pediatric cases are exceptional but documented.
Clinical Features of Malignant Atrophic Papulosis
Cutaneous Lesions
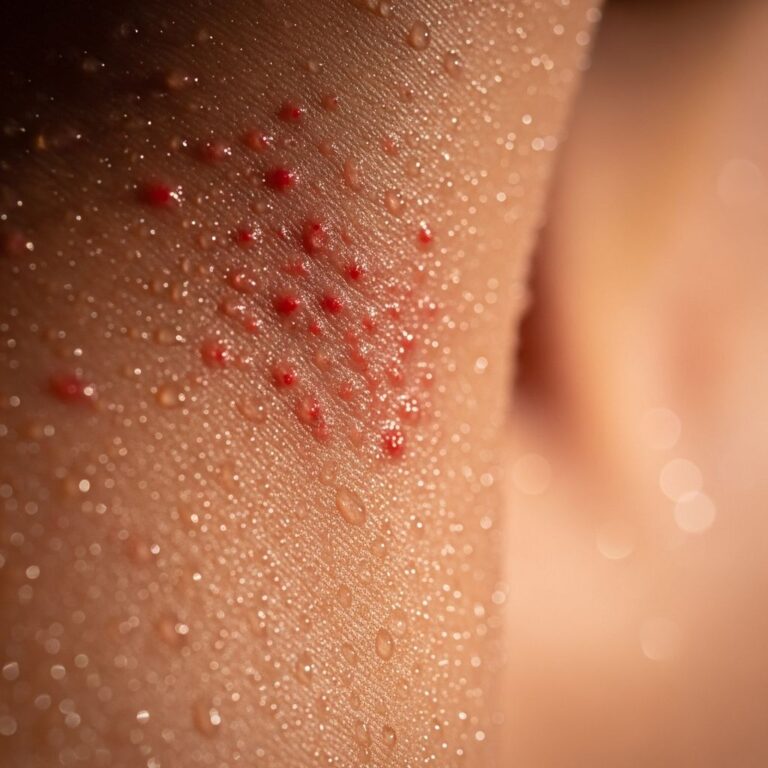
The hallmark of MAP is distinctive skin lesions appearing on the trunk, proximal extremities, neck, and scalp, sparing the face, palms, and soles. Lesions begin as 2-5 mm pink, red, or skin-colored papules that evolve over 2-3 weeks into 5-15 mm scars with central porcelain-white atrophy (0.3-1.0 cm) and a thin erythematous or violaceous telangiectatic rim. Early lesions may be macular or papular; mature ones depress centrally with a ‘punched-out’ appearance. They are often asymptomatic but can cause pain or pruritus. New lesions may appear for months to years, with older ones scarring permanently.
- Size: 2-15 mm diameter
- Number: Typically 20-30, up to hundreds
- Distribution: Trunk (most common), arms, legs; spares mucous membranes
- Evolution: Papule → umbilication → white scar with rim (2 weeks)
Systemic Involvement
Systemic manifestations occur in ~70% of cases, often 6 months to 6 years after skin lesions.
- Gastrointestinal (most common, 50-60%): Abdominal pain, diarrhea, perforation, bleeding; sites include small bowel, stomach
- Neurological (20-30%): Strokes, hemiparesis, seizures, meningitis
- Cardiac/Pericardial (15-20%): Pericarditis, effusion, tamponade
- Ocular: Retinal detachments, uveitis
- Other: Pulmonary hypertension, genitourinary, pleuritis
Mortality reaches 50-90% in systemic MAP from peritonitis (61%), CNS events (18%), or pericardial disease (16%).
Pathogenesis of Malignant Atrophic Papulosis
The etiology is unknown but involves vasculopathy (endothelial swelling/proliferation), coagulopathy (thrombi, fibrinogen elevation, platelet aggregation), and inflammation. Triggers may include viral infections, autoimmunity, or genetic predispositions (e.g., antiphospholipid syndrome overlap). No infectious agent is confirmed.
Diagnosis of Malignant Atrophic Papulosis
Diagnosis relies on clinical lesions plus histopathology; no lab test is specific.
Skin Biopsy (Gold Standard)
Classic findings: wedge-shaped dermal infarction/necrosis, thrombotic vasculopathy of deep vessels, endothelial proliferation, minimal inflammation, mucin deposition. Early: perivascular lymphohistiocytic infiltrate, interface dermatitis. Late: sclerosis, atrophy.
| Stage | Histology |
|---|---|
| Early Papule | Lymphohistiocytic infiltrate, mucin, vacuolar interface |
| Mature | Wedge necrosis, thrombi, vasculitis |
| Late Scar | Sclerosis, epidermal atrophy, no rim inflammation |
Differential Diagnosis
- Benign atrophic papulosis (no systemic signs >3 years)
- Cholesterol emboli, embolizing vasculitis, APS
- Lupus, scleroderma overlaps
Investigations
- Biopsy of skin/GI lesions
- CBC, coagulation panel, ANA
- Endoscopy/CT for GI, MRI brain, echo heart
Treatment of Malignant Atrophic Papulosis
No proven therapy exists; treatments are empiric and focus on anticoagulation, immunosuppression, and symptom management.
- Anticoagulation/Antiplatelet: Aspirin, dipyridamole, warfarin, heparin (common first-line)
- Immunosuppressants: Steroids, azathioprine, cyclophosphamide, tacrolimus
- Biologics/Other: IVIG, infliximab, bevacizumab, treprostinil (promising for skin/PH)
Treprostinil showed rapid skin lesion involution in one case. Supportive care for complications (surgery for perforation). Prognosis: Benign form excellent; systemic poor (median survival 1-2 years).
What is the Prognosis for Malignant Atrophic Papulosis?
Cutaneous-limited: Excellent, lesions scar but stabilize. Systemic: Guarded, with high mortality despite treatment. Early intervention may alter course; registries are advocated. Follow-up: Twice yearly for 7 years, then annually.
Frequently Asked Questions
Is malignant atrophic papulosis contagious?
No, MAP is not infectious or contagious; it is a primary vasculopathy.
Can malignant atrophic papulosis be cured?
There is no cure, but cutaneous forms self-limit, and treatments may control systemic disease.
How is malignant atrophic papulosis diagnosed?
By characteristic skin lesions and confirmatory biopsy showing thrombotic vasculopathy.
What causes the porcelain-white centers in MAP lesions?
They result from dermal infarction and collagen necrosis due to vascular thrombosis.
Is malignant atrophic papulosis associated with other diseases?
Yes, overlaps with lupus, scleroderma, APS.
References
- Malignant Atrophic Papulosis Is Challenging to Diagnose, Treat — The Rheumatologist. 2017-01-01. https://www.the-rheumatologist.org/article/malignant-atrophic-papulosis-is-challenging-to-diagnose-treat/?singlepage=1
- Malignant atrophic papulosis — PubMed/NCBI. 2007-08-01. https://pubmed.ncbi.nlm.nih.gov/17692056/
- Atrophic Papulosis — Karger Publishers (Dermatology). 2023-03-01. https://karger.com/drm/article/239/2/177/841856/Atrophic-Papulosis
- Malignant atrophic papulosis — VisualDx. 2024-01-01. https://www.visualdx.com/visualdx/diagnosis/malignant+atrophic+papulosis?diagnosisId=51146&moduleId=101
- Malignant Atrophic Papulosis — MalaCards. 2024-01-01. https://www.malacards.org/card/malignant_atrophic_papulosis
- Malignant Atrophic Papulosis — StatPearls/NCBI Bookshelf. 2023-07-17. https://www.ncbi.nlm.nih.gov/books/NBK544329/
Similar Articles






Read full bio of medha deb