Marfan Syndrome: Key Facts on Causes, Symptoms & Care
Genetic connective tissue disorder affecting skeleton, eyes, heart, and skin with stretch marks and high stature.

Marfan syndrome is an autosomal dominant inherited disorder of connective tissue due to a defective gene on chromosome 15 producing abnormal fibrillin-1, a glycoprotein essential for elastic fiber formation in connective tissues.
It affects multiple organ systems, primarily the skeleton, eyes, heart and aorta, and skin. Features include unusually tall stature, long limbs and fingers, heart abnormalities, and lens dislocation. Skin manifestations like striae distensae (stretch marks) without weight change or pregnancy are common but not diagnostic alone.
What is the cause of Marfan syndrome?
Marfan syndrome results from mutations in the FBN1 gene on chromosome 15q21.1, encoding fibrillin-1. Over 3,000 mutations identified, mostly missense, leading to haploinsufficiency or dominant-negative effects.
Fibrillin-1 forms microfibrils in elastic fibers and regulates TGF-β signaling. Mutations cause microfibril dysfunction, excessive TGF-β activation, and tissue weakness in aorta, skeleton, eyes, and skin.
Inheritance is autosomal dominant with 75% family history and 25% de novo mutations. Variable expressivity and age-dependent penetrance occur due to allelic heterogeneity.
Who gets Marfan syndrome?
Prevalence is 2–3 per 10,000 worldwide, equal in males and females, all ethnicities. No predilection by race or geography.
- Affects all ages; diagnosis often in childhood/adolescence via growth patterns or complications.
- Family history in ~75%; sporadic cases from new mutations.
What are the clinical features of Marfan syndrome?
Systemic involvement varies; Ghent-2 criteria guide diagnosis. Key systems:
Skeletal features
Tall stature (above 95th percentile), disproportionately long limbs (arm span/height >1.05), arachnodactyly (long fingers, positive wrist/thumb signs).
- Chest deformities: pectus excavatum/carinate (requiring surgery if symptomatic).
- Spinal: scoliosis/kyphosis (50–60%).
- Foot: pes planus, dural ectasia.
- Facial: dolichocephaly, malar hypoplasia, retrognathia, high arched palate, dental crowding.
Ocular features
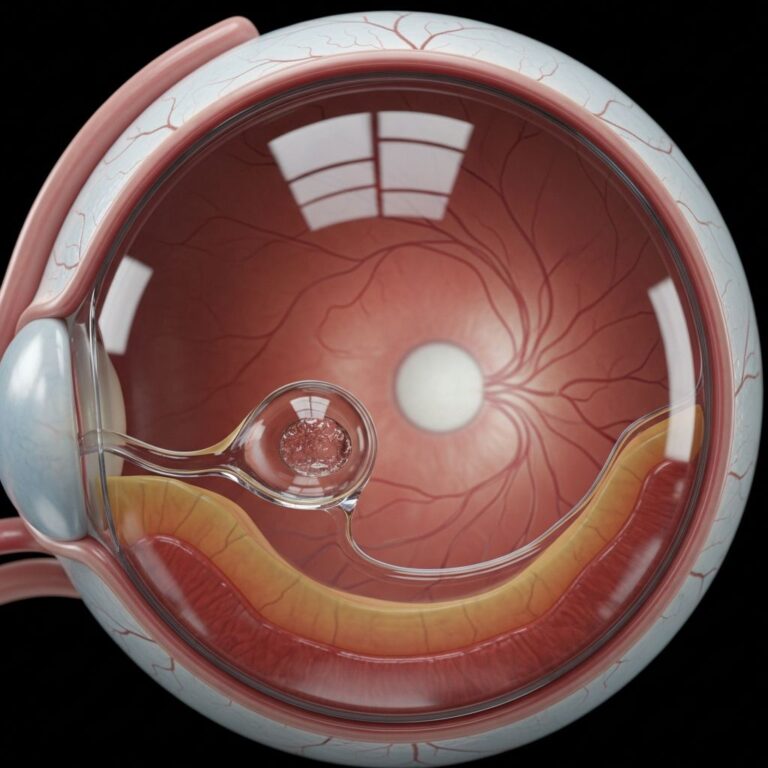
Ectopia lentis (subluxation/dislocation) in 50–80%, typically bilateral, superotemporal.
- Myopia (70%), retinal detachment risk.
- Early glaucoma/cataracts.
Cardiovascular features
Aortic root dilatation/dissection (most life-threatening), mitral valve prolapse (60%).
- Progressive aortic aneurysm; sudden death risk if untreated.
- Family history of aortic dissection flags urgency.
Skin features
Striae distensae (92% vs 61% controls) on shoulders, lower back, thighs, hips, buttocks—without weight gain/pregnancy. Due to elastic fiber defects.
- Hyperextensible, velvety thin skin.
- Poor wound healing: hypertrophic/atrophic scars (46% vs 21% controls).
- Hernias (inguinal/umbilical).
- Rare: vasculitic-like rashes, mottled skin, livedo reticularis.
Striae specificity high (84%) in unusual sites (not buttocks/hips/thighs).
Other features
- Lungs: spontaneous pneumothorax, emphysema.
- Nervous: dural ectasia (lumbosacral pain).
- Dental: high palate, crowding.
Diagnosis of Marfan syndrome
Revised Ghent-2 criteria (2010) integrate clinical, echocardiographic, genetic findings. No single test; systemic score ≥7 points.
| Feature | Criteria |
|---|---|
| Aorta | √Z ≥2 (age <20), ≥3 (>20); or dissection |
| Ectopia lentis | With FBN1 mutation |
| FBN1 mutation | Known aortic/lens causality + systemic ≥7 or aorta ≥Z above |
| Systemic score | Wrist AND thumb 3; wrist OR thumb 1 Pectus carinatum 2; excavatum/major surgery 1 Face ≥3/5 features 2; 2 1 Pes planus 1; pneumothorax 1; dural ectasia 2 Protrusio acetabuli 1; scoliosis/kyphosis 1 Arm/height ≥1.05 1; dolichocephaly 1 Striae 1; severe myopia/hypotonia/malocclusion 1 each |
No family history: Aorta + ectopia OR aorta + FBN1 + systemic ≥7 OR ectopia + FBN1 causality OR systemic ≥7 + minus 1.
Family history: Ectopia OR aorta OR systemic ≥7 OR FBN1.
Genetic testing confirms FBN1 variants (coverage 70–93%). Imaging: echocardiography (aorta), slit-lamp (lens), MRI (dural ectasia).
Differentiate from MASS phenotype (mitral prolapse, skin/striae, skeletal; milder aorta), Loeys-Dietz, Shprintzen-Goldberg.
Management of Marfan syndrome
Multidisciplinary: cardiologist, ophthalmologist, geneticist, orthopaedist, dermatologist. Lifelong surveillance.
Cardiovascular
- β-blockers (atenolol) from diagnosis; losartan for TGF-β modulation.
- Echocardiogram 6-monthly if Z≥3; surgery at Z≥5 or rapid growth.
- Avoid contact sports, isometric exercise; isometrics OK if monitored.
Ophthalmic
- Annual slit-lamp; correct refractive errors; lens surgery if vision impaired.
Skeletal
- Orthotics/bracing for scoliosis/foot; surgery for severe pectus/scoliosis.
- Physiotherapy for joint laxity.
Skin
Striae untreated (cosmetic); optimize wound care/sutures for healing. Hernia repair as needed.
Pregnancy/family planning
High-risk; preconception counseling, monthly fetal echo. Avoid if aorta >45mm.
Prognosis
Improved to normal lifespan with monitoring; aortic dissection main risk.
Frequently Asked Questions
What is Marfan syndrome?
Autosomal dominant connective tissue disorder from FBN1 mutations affecting skeleton, eyes, heart, skin.
Is Marfan syndrome curable?
No cure; managed with medications, surveillance, surgery to prevent complications.
Can Marfan syndrome be prevented?
Genetic counseling/testing for families; early diagnosis key. No prevention for de novo cases.
Are stretch marks diagnostic for Marfan?
Common (92%) especially unusual sites (specificity 84%), but part of systemic score, not alone.
What sports are safe for Marfan patients?
Low-impact: walking, swimming, cycling. Avoid weightlifting, contact/high-static sports.
References
- Marfan Syndrome Cutaneous Manifestations — DrOracle.ai. 2024. https://www.droracle.ai/articles/139608/what-are-the-cutaneous-manifestations-of-marfan-syndrome
- Marfan syndrome (MFS) mimicking cutaneous vasculitis — PMC/NCBI. 2023-07-01. https://pmc.ncbi.nlm.nih.gov/articles/PMC10308279/
- Elastosis Perforans Serpiginosa Is Not a Cutaneous Manifestation of Marfan Syndrome — Cureus. 2024. https://www.cureus.com/articles/387598-elastosis-perforans-serpiginosa-is-not-a-cutaneous-manifestation-of-marfan-syndrome.pdf
- A case-control study of cutaneous signs in adult patients with Marfan syndrome — PubMed/J Am Acad Dermatol. 2010-12-01. https://pubmed.ncbi.nlm.nih.gov/21112669/
- Marfan syndrome — DermNet NZ. 2024. https://dermnetnz.org/topics/marfan-syndrome
- Signs and Symptoms: a Deadly Secret? — Marfan Trust. 2024. https://www.marfantrust.org/articles/signs-and-symptoms-a-deadly-secret
- Marfan syndrome – Symptoms and causes — Mayo Clinic. 2024-10-20. https://www.mayoclinic.org/diseases-conditions/marfan-syndrome/symptoms-causes/syc-20350782
Similar Articles






Read full bio of Sneha Tete