Non-Langerhans Cell Histiocytosis: A Comprehensive Guide
Comprehensive guide to non-Langerhans cell histiocytosis: causes, types, symptoms, diagnosis, and treatments for these rare skin disorders.

Non-Langerhans cell histiocytosis
Author: Reviewed by Dermatologists
Non-Langerhans cell histiocytosis refers to a diverse group of rare disorders characterised by the abnormal accumulation and proliferation of
histiocytes
—specialised cells from the mononuclear phagocyte system that are distinct from Langerhans cells. These conditions primarily manifest as reddish-brown or yellowish papulonodular skin lesions but can involve internal organs such as the liver, lungs, kidneys, and eyes. Unlike Langerhans cell histiocytosis (LCH), which involves cells expressing CD1a and S100 with Birbeck granules, non-LCH histiocytes lack these markers and arise from dermal dendritic cells or monocyte-macrophage lineages.The term ‘non-Langerhans cell histiocytosis’ (also known as class II histiocytosis, non-X histiocytosis, or histiocytosis of mononuclear phagocytes other than Langerhans cells) encompasses both hereditary and acquired diseases, often with a tendency for spontaneous regression in infantile forms but progression in adult-onset cases. Recent classifications by the Histiocyte Society divide them into Class IIa (dermal dendritic cell-related) and Class IIb (monocyte-macrophage related), aiding in understanding pathogenesis and guiding therapy. This article details the subgroups, specific entities, clinical features, histopathology, diagnosis, and management.
What is non-Langerhans cell histiocytosis?
Histiocytes are tissue macrophages derived from bone marrow monocytes, functioning in phagocytosis, antigen presentation, and inflammation. In non-LCH, clonal or reactive proliferation of these cells leads to granulomatous lesions. Bone marrow and blood monocytes give rise to macrophages, histiocytes, indeterminate cells, and dendritic cells, with non-LCH typically showing lipid-laden (xanthomatized) histiocytes, Touton giant cells, and minimal phagocytosis compared to LCH.
Clinically, these disorders are stratified into: (1) predominantly cutaneous forms (e.g., juvenile xanthogranuloma in infants, self-healing); (2) skin-involved systemic diseases (e.g., Erdheim-Chester disease); and (3) extracutaneous predominant (e.g., Rosai-Dorfman disease with secondary skin involvement). Infancy-onset lesions are often papulonodular and regress spontaneously, while adult forms present as plaques and may be aggressive. Genetic mutations (e.g., in MAPK/ERK pathway for some) and paraprotein associations in others drive pathogenesis.
Who gets non-Langerhans cell histiocytosis?
Non-LCH affects all ages, with specific entities showing preferences: juvenile xanthogranuloma (JXG) predominantly infants under 1 year (67% solitary lesions); generalized eruptive histiocytosis (GEH) in young adults; and multicentric reticulohistiocytosis in middle-aged women. Incidence is rare, estimated at 1-5 per million, higher in Caucasians for some types. Risk factors include genetic predispositions (hereditary progressive mucinous histiocytosis) and associations with paraproteinemias or myelodysplastic syndromes.
What causes non-Langerhans cell histiocytosis?
Aetiology is multifactorial: most cases are sporadic, but clonal mutations (e.g., BRAF, MAP2K1 in JXG spectrum) suggest neoplastic processes in some. In necrobiotic xanthogranuloma (NXG), immunoglobulin-lipid complexes trigger foreign-body reactions, linked to paraproteinemia. Hereditary forms involve X-linked patterns. Reactive hyperplasia from infections, autoimmunity, or lipids occurs in others. Dermal dendrocytes, factor XIIIa-positive precursors, are implicated in Class IIa disorders.
What are the clinical features of non-Langerhans cell histiocytosis?
Skin lesions are hallmark: reddish-brown or yellow papules, nodules, or plaques, often on trunk, head, and flexures. Systemic involvement varies—ocular in JXG (iris, glaucoma risk), pulmonary in ECD, lymphadenopathy in Rosai-Dorfman.
Class IIa histiocytosis (dermal dendritic cell-related)
- Juvenile xanthogranuloma (JXG): Most common; orange-red to yellow papules/nodules (2-5mm), upper body; solitary (67%) or multiple; self-resolves in 90%; eye involvement in 0.4%.
- Benign cephalic histiocytosis (BCH): Infants; red-brown papules on head/neck; resolves by age 3.
- Generalized eruptive histiocytosis (GEH): Adults/children; hundreds of pink-red papules (3-10mm) in crops, symmetric; mucous membranes spared in kids.
- Papular xanthoma (PX): Foam/Touton cells; yellow papules without primitive phase.
- Progressive nodular histiocytosis: Nodular plaques; persistent.
Class IIb histiocytosis (monocyte-macrophage related)
Hereditary
- Hereditary progressive mucinous histiocytosis: Women; mucin-laden histiocytes on limbs; X-linked.
Acquired
- Xanthoma disseminatum: Yellow-red papules, mucous membranes; diabetes insipidus.
- Reticulohistiocytoma cutis/reticulohistiocytosis: Solitary/cutaneous nodules.
- Multicentric reticulohistiocytosis: Skin nodules + arthritis; paraprotein link.
- Necrobiotic xanthogranuloma (NXG): Periorbital plaques with ulceration; paraproteinemia, eye/lymphoma risk.
- Erdheim-Chester disease (ECD): Adult; xanthelasma-like, bone/systemic.
- Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman): Cervical nodes, skin; emperipolesis.
- Indeterminate cell histiocytosis: S100+ CD1a-; aggressive.
How is non-Langerhans cell histiocytosis diagnosed?
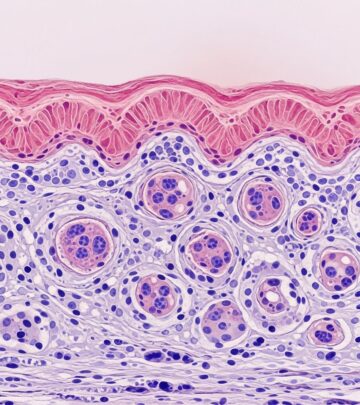
Diagnosis relies on clinicopathologic correlation: full-thickness skin biopsy showing non-Langerhans histiocytes (CD68+, CD1a-, S100 variable). Early lesions: monomorphic histiocytes; mature: foam cells, Touton giants (wreath-like nuclei, lipid rim), fibrosis. Immunohistochemistry: Class IIa (factor XIIIa+, CD163+); Class IIb (CD163+, lysozyme+). Ultrastructure: comma-shaped bodies in JXG, lipid vacuoles. Imaging (MRI/PET for ECD), labs (paraproteins), and exclusion of lipids/metabolic disorders essential.
| Entity | Key Histology | Immuno |
|---|---|---|
| JXG | Touton giants, foam cells | CD68+, FXIIIa+, CD1a- |
| NXG | Necrobiosis, giants | CD68+, paraprotein assoc. |
| ECD | Foamy histiocytes, fibrosis | BRAF mut. possible |
What is the treatment for non-Langerhans cell histiocytosis?
Treatment is entity-specific: observation for self-limited (JXG, BCH); excision for solitary lesions. Systemic: corticosteroids, methotrexate, cladribine for progressive/systemic (ECD, multicentric reticulohistiocytosis). Targeted: BRAF inhibitors (vemurafenib) for mutated cases. Imiquimod, phototherapy for cutaneous; IVIG, rituximab for NXG/paraprotein. Prognosis excellent for cutaneous infantile forms (90% resolve); guarded for systemic (mortality 20-30% in ECD). Multidisciplinary care vital.
Frequently asked questions (FAQs) about non-Langerhans cell histiocytosis
What is the difference between Langerhans and non-Langerhans cell histiocytosis?
LCH features CD1a+/S100+ cells with Birbeck granules, often aggressive/systemic; non-LCH lacks these, from dermal dendrocytes/macrophages, more cutaneous/benign.
Is juvenile xanthogranuloma a type of cancer?
No, JXG is benign/self-healing; rare atypical cases may have clonal mutations but excellent prognosis.
Can non-LCH affect internal organs?
Yes, e.g., eyes in JXG, bones/lungs in ECD, nodes in Rosai-Dorfman; monitor with imaging.
How is biopsy used in diagnosis?
Shows Touton giants, foam cells; immuno excludes LCH (CD1a-).
What is the prognosis for adults with non-LCH?
Variable; cutaneous good, systemic (ECD) poorer, responsive to targeted therapy.
References
- Cutaneous Syndromes of Non-X Histiocytosis — JAMA Dermatology. 1986. https://jamanetwork.com/journals/jamadermatology/fullarticle/542583
- Uncommon histiocytic disorders: the non-Langerhans cell histiocytoses — PubMed (Clin Dermatol). 2004-11-24. https://pubmed.ncbi.nlm.nih.gov/15547923/
- Non-Langerhans cell histiocytosis — DermNet NZ. Recent. https://dermnetnz.org/topics/non-langerhans-cell-histiocytosis
- Non-Langerhans Cell Histiocytosis — Plastic Surgery Key. Recent. https://plasticsurgerykey.com/non-langerhans-cell-histiocytosis/
- Histiocytoses – Overview — Perri Dermatology. Recent. https://perridermatology.com/dr-perris-blog/histiocytoses-overview/
- Unraveling cutaneous histiocytosis: insights into histology — Frontiers in Medicine. 2025. https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2025.1585815/full
Similar Articles

Read full bio of medha deb





