Rubinstein-Taybi Syndrome
Comprehensive guide to Rubinstein-Taybi syndrome: genetics, symptoms, diagnosis, and multidisciplinary management strategies.

Rubinstein-Taybi syndrome (RTS), also known as broad thumb-hallux syndrome, is a rare multisystem genetic disorder primarily caused by pathogenic variants in the CREBBP (RTS1) or EP300 (RTS2) genes. These genes encode proteins involved in transcription regulation and histone acetylation, leading to a phenotypic spectrum that includes intellectual disability, distinctive craniofacial features, broad thumbs and halluces, growth abnormalities, and dermatological issues like keloid scarring.
What is Rubinstein-Taybi syndrome?
RTS affects approximately 1 in 100,000–125,000 live births, presenting with a combination of physical, cognitive, and behavioral characteristics. First described in 1963 by Jack Rubinstein and Hooshang Taybi, the syndrome manifests variably from mild to severe forms. Core features include postnatal growth retardation, moderate to severe intellectual disability (average IQ 25–79), and broad, often angulated thumbs and first toes. Additional common traits encompass craniofacial dysmorphism, cardiac anomalies, eye abnormalities, feeding difficulties, and a predisposition to tumors and excessive scarring.
The condition arises from disrupted chromatin remodeling, impairing gene expression critical for development. While most cases are de novo mutations, autosomal dominant inheritance occurs rarely. Phenotypic overlap exists between RTS1 (50–60% of cases, CREBBP) and RTS2 (3–8%, EP300), though RTS1 tends to show more severe intellectual impairment and microcephaly.
Who gets Rubinstein-Taybi syndrome (Epidemiology)?
RTS is equally prevalent in males and females, with no strong ethnic predisposition reported. Incidence estimates vary due to underdiagnosis, but consensus places it at 1:100,000–125,000. Diagnosis often occurs in infancy via dysmorphic features, though milder cases may be identified later. Prenatal growth is typically normal, but postnatal failure to thrive is universal, leading to short stature below the third percentile in most.
Geographic variation in recognition stems from access to genetic testing; international expert consensus highlights diagnostic heterogeneity globally.
Clinical features
RTS exhibits a broad phenotypic spectrum. Key features are categorized below.
Craniofacial features
- Highly arched eyebrows with long eyelashes
- Downslanting palpebral fissures
- Prominent, beaked nose with broad nasal bridge
- Low-set retrognathia (receding chin)
- High-arched or narrow palate
- Microcephaly (in ~80% of RTS1 cases)
- Low posterior hairline and thick scalp hair
These traits contribute to a characteristic facial gestalt recognizable by experts.
Limb anomalies
- Broad, short thumbs and halluces, often angulated
- Brachydactyly (short fingers/toes)
- Clinodactyly and syndactyly in some
Surgical correction may be considered for severe functional impairment.
Growth and development
- Postnatal short stature (<3rd percentile)
- Failure to thrive and feeding difficulties (dysphagia)
- Motor delays: hypotonia, unsteady gait, hyperreflexia
- Cognitive delays: intellectual disability, speech impairment
Obesity may develop in later childhood despite early poor weight gain.
Cardiac and other systemic
- Congenital heart defects in ~30% (e.g., VSD, PDA, murmurs)
- Renal anomalies (e.g., hydronephrosis)
- Eye issues: strabismus, glaucoma, cataracts, nasolacrimal duct obstruction
- Dental: talon cusps, crowded teeth, hypodontia
- Genitourinary: cryptorchidism, hypospadias
Increased tumor risk includes pilomatricomas, meningiomas, and keloids.
Dermatological features
Skin manifestations are prominent in RTS, including:
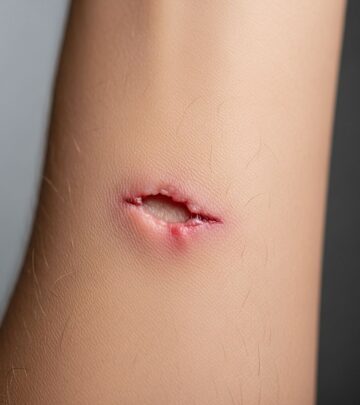
- Keloid scarring: Atraumatic, hypertrophic scars post-minor trauma or surgery (pathognomonic)
- Hypertrichosis and hirsutism
- Pilomatricomas (benign hair matrix tumors)
- Fungal nail infections, ingrown toenails
- Capillary malformations (e.g., forehead “strawberry mark”)
Keloids arise due to dysregulated fibroblast activity from CBP/p300 dysfunction.
Diagnosis
Clinical diagnosis relies on cardinal and supportive criteria (adapted from international consensus).
| Cardinal Features (Required) | Supportive Features |
|---|---|
| Face (≥3/6): Arched eyebrows, downslant palpebral fissures, beaked nose, grimacing smile, low columella, small alae nasi | Maternal pre-eclampsia |
| Thumbs/halluces: Broad/angulated | Keloids |
| Intellectual disability | Hypertrichosis |
Molecular confirmation via sequencing of CREBBP (16p13.3) and EP300 (22q13.2) detects ~70–80% of cases; deletions, point mutations, or intragenic rearrangements. Array CGH or MLPA for larger variants. Negative testing does not exclude if clinical score is high.
Genetic testing and counselling
Indications: Dysmorphic features + broad thumbs/halluces + developmental delay. Trio exome sequencing recommended if single-gene negative. Variants are mostly de novo (99%), but parental mosaicism possible. Recurrence risk <1%, but offer prenatal testing. Multidisciplinary counseling addresses cognitive, medical, and psychosocial implications.
Differential diagnosis
- Other overgrowth syndromes: Cornelia de Lange, Weaver, Simpson-Golabi-Behmel
- Floating-Harbor syndrome (short stature, facial features)
- Deletion 2q37 (brachydactyly)
- Trisomy 18 mosaicism
Distinctive RTS gestalt and genetic testing differentiate.
Disease severity and prognosis
Severity varies: Mild (IQ >50, independent living possible) to profound disability. Life expectancy is near-normal with management, though respiratory infections, aspiration, and tumors pose risks. Behavioral issues (anxiety, ADHD, autism traits) impact quality of life. Early intervention improves outcomes.
Management
Multidisciplinary, lifelong care per international consensus.
- Growth/Endocrinology: Screen for GH deficiency if growth falters; hypoglycemia in neonates.
- Cardiac: Echocardiogram at diagnosis; monitor murmurs.
- Eye/Hearing: Annual exams; treat strabismus, glaucoma.
- Dental: Specialist care for talon cusps, hypodontia.
- Skin: Minimize trauma to prevent keloids; silicone gels post-surgery.
- Developmental: Physiotherapy, speech therapy, special education.
- Behavioral: CBT for anxiety, antipsychotics if needed.
- Tumors: Screen for pilomatricomas, brain tumors.
Surgical interventions for thumbs/halluces only if functional.
Treatment of complications
- Keloids: Intralesional steroids, cryotherapy; prevent via atraumatic techniques.
- Constipation: Laxatives, high-fiber diet (common due to hypotonia).
- Seizures: EEG/antiepileptics if present (~20%).
- Obesity: Diet/exercise counseling.
Sexual health education essential for adolescents.
Prevention
No primary prevention; genetic counseling for families. Prenatal diagnosis via CVS/amnio if parental variant known.
Clinical trials
Ongoing research into histone deacetylase inhibitors and targeted therapies for CBP/p300 dysfunction. Support groups like Jack Rubinstein Foundation facilitate trial access.
Patient support organizations
- Rubinstein-Taybi Syndrome Support Group (UK)
- Jack Rubinstein Foundation (US)
- ERN-ITHACA (Europe)
Frequently Asked Questions
What causes Rubinstein-Taybi syndrome?
RTS results from mutations in CREBBP (RTS1, ~60%) or EP300 (RTS2, ~5–10%), disrupting epigenetic regulation.
Is RTS inherited?
Mostly de novo; rare autosomal dominant inheritance. Recurrence risk low (<1%).
Can keloid scarring be prevented?
Yes, via careful wound care, silicone sheets, and pressure garments post-trauma/surgery.
What is the life expectancy?
Near-normal with vigilant care; monitor for cardiac, respiratory, and tumor complications.
Are there growth charts specific to RTS?
Yes, syndrome-specific charts aid monitoring.
References
- Diagnosis and management in Rubinstein-Taybi syndrome — Nguyen-Tu MS et al. Nat Rev Endocrinol. 2024. https://pmc.ncbi.nlm.nih.gov/articles/PMC11137475/
- Rubinstein-Taybi syndrome — MedlinePlus Genetics. U.S. National Library of Medicine. Updated 2023. https://medlineplus.gov/genetics/condition/rubinstein-taybi-syndrome/
- Rubinstein-Taybi Syndrome — NORD (National Organization for Rare Disorders). Updated 2023. https://rarediseases.org/rare-diseases/rubinstein-taybi-syndrome/
- Rubinstein-Taybi syndrome — HealthFinder (Florida Dept. of Health). Accessed 2026. https://quality.healthfinder.fl.gov/health-encyclopedia/HIE/1/001249
- Rubinstein-Taybi Syndrome — DermNet NZ. Updated 2023. https://dermnetnz.org/topics/rubinstein-taybi-syndrome
Similar Articles
Read full bio of medha deb





