Schnitzler Syndrome: Guide To Symptoms, Diagnosis And Treatment
Rare autoinflammatory disorder with chronic urticaria, IgM gammopathy, fever, and bone pain—insights into diagnosis and IL-1 blockade therapy.

Schnitzler Syndrome
Schnitzler syndrome is a rare, late-onset acquired autoinflammatory disorder defined by the combination of chronic urticarial rash and monoclonal IgM gammopathy, often accompanied by systemic symptoms such as fever, arthralgia, bone pain, and elevated inflammatory markers.
What is Schnitzler Syndrome?
Schnitzler syndrome, first described in 1972 by French dermatologist Philippe Schnitzler, represents an acquired form of autoinflammatory disease resembling cryopyrin-associated periodic syndromes (CAPS) but occurring in adults without NLRP3 mutations. It is characterized by immunoglobulin M (IgM) monoclonal gammopathy of undetermined significance (MGUS) alongside a distinctive non-pruritic urticarial eruption and constitutional symptoms. The syndrome affects middle-aged to older adults, predominantly males, with an estimated prevalence under 1 in 1,000,000, though underdiagnosis is common due to its multisystem presentation involving dermatology, rheumatology, and hematology. Pathogenetically, it involves dysregulated inflammasome activation leading to overproduction of interleukin-1β (IL-1β), driving neutrophilic inflammation, NETosis, and clinical manifestations. Unlike classic urticaria, the rash resists antihistamines, and the paraprotein does not directly cause skin lesions, as deep sequencing shows no shared B-cell clonality. Patients often experience diagnostic delays spanning years, presenting to multiple specialists before recognition.
Who Gets Schnitzler Syndrome (Epidemiology)?
Schnitzler syndrome typically manifests in individuals over 50 years old, with a mean age at onset around 52–63 years, showing a slight male predominance (male-to-female ratio approximately 1.5–2:1). It is exceedingly rare, with fewer than 300 cases reported worldwide as of recent reviews, though increased awareness may reveal higher incidence. No strong ethnic or geographic predisposition exists, but cases cluster in Europe and North America due to reporting bias. Risk factors remain unclear, but late-onset autoinflammation suggests somatic events or environmental triggers dysregulating innate immunity, akin to low-grade somatic NLRP3 mosaicism in some CAPS-like presentations. Comorbidities include Waldenström macroglobulinaemia in 10–15% of untreated cases, emphasizing the need for vigilant monitoring.
Clinical Features
The hallmark of Schnitzler syndrome is a chronic urticarial rash, present in nearly 100% of cases, often preceding other symptoms by months to years. Systemic involvement affects over 90% of patients, including recurrent fevers, musculoskeletal pain, organomegaly, and haematological anomalies.
Urticaria
The rash consists of recurrent, monomorphic pink-to-red papules or plaques (0.5–5 cm) on the trunk, limbs, and flexures, sparing the face, palms, and soles. Lesions are barely raised, non-pitting, and resolve within 24 hours without scarring or hyperpigmentation, though new crops emerge daily or episodically. Unlike ordinary hives, pruritus is absent or minimal (burning sensation in 20%), and dermographism or vasoconstrictive halos may appear; angioedema is rare. Antihistamines fail to control it, distinguishing it from allergic urticaria.
Arthritis/Arthralgia
Musculoskeletal symptoms occur in 80–90% of patients, manifesting as arthralgia (joint pain) or non-erosive arthritis affecting knees, wrists, and ankles. Bone pain, reported in 50–70%, stems from abnormal remodelling visible on bone scans (hyperostosis, osteosclerosis).
Fever
Intermittent fevers exceeding 38°C affect 85–90%, often with chills and night sweats, lasting hours to days and coinciding with rash flares.
Organomegaly
Lymphadenopathy (60–80%), hepatomegaly (15–20%), and splenomegaly (15%) arise from inflammatory infiltrates.
Haematological Abnormalities
Anaemia (normocytic), leukocytosis/neutrophilia (>10,000/mm³), thrombocytosis, and elevated CRP/ESR are common. IgM paraproteinaemia (kappa light chain predominant) is obligatory.
Other Features
- Neurological: Cranial nerve palsies, sensorineural deafness (10–15%), headache.
- Renal: Renal impairment from AA amyloidosis (5–10% untreated).
- Endocrine: Rare hypertrichosis, hyperpigmentation.
Diagnosis of Schnitzler Syndrome
Diagnosis relies on clinical, laboratory, and histopathological correlation, using the Strasbourg criteria (2012), which offer 95% sensitivity and specificity.
| Strasbourg Diagnostic Criteria | |
|---|---|
| Obligate (Both Required) | • Chronic urticarial rash • Monoclonal IgM gammopathy (or IgG in atypical cases) |
| Minor (≥2 Required for Diagnosis) | • Recurrent fever >38°C • Objective bone remodelling (scan/BM abnormalities) ± pain • Neutrophilic urticarial dermatosis on biopsy • Leukocytosis/neutrophilia >10,000/mm³ or CRP >30 mg/L • Monoclonal IgG gammopathy (if IgM absent) |
Laboratory confirmation includes serum protein electrophoresis/immunofixation for IgM MGUS (median 10–30 g/L), raised CRP (50–100 mg/L), and exclusion of differentials.
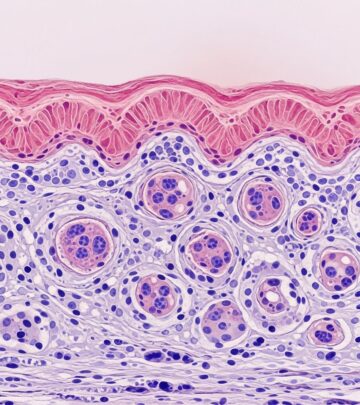
Skin Biopsy
Shows neutrophilic urticarial dermatosis: dense dermal neutrophilic infiltrate without vasculitis, leukocytoclasia, or significant eosinophils; epitheliotropism around adnexa suggestive.
Bone Imaging
99mTc bone scintigraphy reveals increased uptake at periarticular sites.
Differential Diagnosis
- Urticarial Vasculitis: Hypocomplementaemic, longer-lasting weals, pruritus, vasculitis on biopsy.
- CAPS (FCAS/MWS/NOMID): Onset <6 months, NLRP3 mutations, no gammopathy.
- Adult-Onset Still’s: Evanescent salmon rash, sore throat, no gammopathy.
- Systemic Mastocytosis: Darier sign, KIT mutations, tryptase elevation.
- Waldenström Macroglobulinaemia: High-volume IgM, lymphoplasmacytic lymphoma.
Management and Prognosis
Schnitzler syndrome responds dramatically to IL-1 inhibitors, confirming its autoinflammatory nature.
- First-Line: Anakinra (100 mg SC daily): Complete remission in 80–90% within days; safe long-term, though daily injections needed.
- Alternatives: Canakinumab (q4–8 weeks), rilonacept; off-label.
- Steroids/Immunosuppressants: Partial response; not recommended long-term.
Prognosis is favourable with treatment: no progression to lymphoma in IL-1 blocker era (vs. 15% untreated); monitor for amyloidosis, Waldenström. Discontinuation leads to relapse.
Investigations
| Investigation | Findings |
|---|---|
| Serum Protein Electrophoresis | IgM kappa/lambda MGUS |
| Histopathology | Neutrophilic dermal infiltrate |
| Bloods (FBC, CRP, ESR) | Neutrophilia, anaemia, ↑acute phase |
| Bone Scan | Hyperfixation (bone remodelling) |
| Echo/CT | Lymphadenopathy, organomegaly |
Frequently Asked Questions
What causes Schnitzler syndrome?
An acquired inflammasome dysregulation, likely IL-1β overproduction from mast cells/neutrophils, without identifiable genetic mutations; serum factors enhance NETosis.
Is the rash itchy?
Rarely; unlike hives, it’s non-pruritic or mildly burning, unresponsive to antihistamines.
How is it treated?
IL-1 blockade with anakinra achieves rapid, sustained remission.
Does it turn into cancer?
10–15% progress to Waldenström if untreated; treatment prevents this.
Who should I see?
Dermatologist, rheumatologist, or haematologist for multidisciplinary evaluation.
References
- Schnitzler Syndrome: Insights into Its Pathogenesis, Clinical Features, Diagnosis and Novel Therapeutic Approaches — de Koning HD et al. J Clin Med. 2024-07-15. https://pmc.ncbi.nlm.nih.gov/articles/PMC11202231/
- Schnitzler Syndrome — National Organization for Rare Disorders (NORD). 2023-05-12. https://rarediseases.org/rare-diseases/schnitzler-syndrome/
- Schnitzler syndrome — DermNet NZ. 2024-02-20. https://dermnetnz.org/topics/schnitzler-syndrome
- Schnitzler’s Syndrome: A Diagnostic Consideration in Evaluating the Recurrent Urticarial Rash — Goyal A et al. Cureus. 2021-06-28. https://pmc.ncbi.nlm.nih.gov/articles/PMC8256912/
Similar Articles

Read full bio of Sneha Tete



