Schöpf-Schulz-Passarge Syndrome: Diagnosis & Care
Rare ectodermal dysplasia featuring palmoplantar keratoderma, eyelid cysts, hypodontia, and nail dystrophy with genetic WNT10A mutations.

Schöpf-Schulz-Passarge syndrome (SSPS) is a rare form of ectodermal dysplasia characterized by abnormalities in ectodermally derived structures including skin, hair, nails, and teeth. It typically presents with palmoplantar keratoderma, multiple eyelid apocrine hidrocystomas, hypodontia, hypotrichosis, and nail dystrophy.
Who gets Schöpf-Schulz-Passarge syndrome (SSPS)?
SSPS is an extremely rare autosomal recessive genetic disorder, with fewer than 50 cases reported worldwide. It affects both males and females equally and has been documented in various ethnic groups, though consanguinity is noted in some families, increasing risk due to homozygous mutations. Symptoms often begin in childhood or adolescence but are frequently diagnosed in adulthood, with a mean age of 64 years in reported cases, as milder features may go unnoticed initially.
Heterozygous carriers of the causative gene mutation may exhibit milder abnormalities such as sparse hair, nail changes, or dental issues, suggesting incomplete penetrance. The condition’s rarity poses challenges for early recognition, particularly in isolated or mild presentations.
Causes
SSPS results from biallelic (homozygous or compound heterozygous) mutations in the WNT10A gene located on chromosome 2q35. This gene encodes a signaling protein crucial for cell-cell interactions during embryogenesis, particularly in tooth and hair follicle development through regulation of the β-catenin degradation complex. Over 16 distinct WNT10A mutations have been identified, with considerable clinical overlap with odonto-onycho-dermal dysplasia (OODD).
Inheritance is primarily autosomal recessive, requiring two mutated alleles (one from each parent). However, pseudodominant transmission can occur in consanguineous families or heterozygous carriers with localized disease expression. Sporadic cases have also been reported. The syndrome was first described in 1971 by German dermatologists Schöpf, Schulz, and Passarge in two sisters from a consanguineous marriage.
Clinical features
Clinical manifestations of SSPS primarily affect ectodermal structures and evolve progressively from childhood onward, becoming more prominent in adulthood. Cutaneous features include:
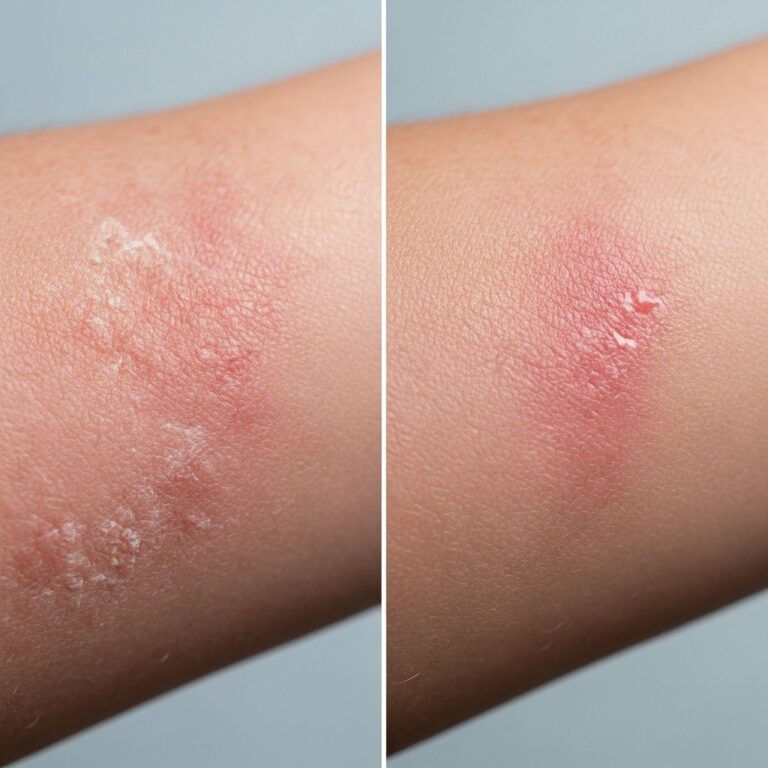
- Palmoplantar keratoderma: Punctate or diffuse symmetric hyperkeratosis on palms and soles, often starting around age 12, with histologic features of eccrine syringofibroadenoma in nearly half of cases.
- Multiple eyelid and periocular apocrine hidrocystomas: The most consistent feature (100% of cases), appearing as translucent cysts on eyelids and periocular skin in adulthood; they enlarge and multiply over time.
- Hypotrichosis: Sparse scalp hair with fine scaling; eyebrows and eyelashes may be thin or absent.
- Nail dystrophy: Fragile, ridged, or dystrophic nails, often anonychia or onycholysis.
- Telangiectatic facial papules or rosacea-like changes: Common on the face.
Non-cutaneous features encompass:
- Hypodontia or oligodontia: Missing teeth, conical or peg-shaped permanent teeth; deciduous teeth often normal initially.
- Smooth tongue: Loss of filiform and fungiform papillae, overlapping with OODD.
- Other: Bird-like facies in some, hypoplasia of alveolar ridges, and rare ocular issues like early cataracts or optic atrophy.
Complications
Complications of SSPS include skin fragility and pain from palmoplantar hyperkeratosis, cosmetic disfigurement from enlarging eyelid cysts, and functional issues with mastication due to dental anomalies. Malignant transformation is a concern: palmoplantar keratoderma may develop squamous cell carcinoma, and adnexal tumors such as eccrine poroma, basal cell carcinoma, or follicular infundibulum tumors have been reported.
Progressive worsening with age affects quality of life, with secondary teeth failing to erupt or being malformed. Associated syringofibroadenomas can cause discomfort, and rare systemic features like arteriosclerotic fundi add to morbidity.
Diagnosis
Diagnosis of SSPS is clinical, supported by characteristic triad of palmoplantar keratoderma, eyelid hidrocystomas, and ectodermal defects (hair, nails, teeth), often confirmed in adulthood. Skin biopsy of eyelid lesions shows apocrine hidrocystomas with decapitation secretion, while palmoplantar biopsies reveal syringofibroadenoma.
Genetic testing for WNT10A mutations is confirmatory, revealing biallelic variants. Differential diagnosis includes other ectodermal dysplasias (e.g., Clouston syndrome, pachyonychia congenita), isolated hidrocystomas, or OODD due to overlapping features. Imaging or dental evaluation aids in assessing hypodontia.
Differential diagnoses
| Condition | Key Distinguishing Features |
|---|---|
| Odonto-onycho-dermal dysplasia (OODD) | Similar WNT10A mutations; lacks eyelid hidrocystomas but shares smooth tongue, nail dystrophy, hypodontia. |
| Ectodermal dysplasia (e.g., hypohidrotic) | Sweat gland involvement absent in SSPS; more anhidrosis. |
| Pachyonychia congenita | Painful keratoderma, oral leukokeratosis; different genetics. |
| Isolated multiple apocrine hidrocystomas | No ectodermal features; sporadic. |
| Clouston syndrome | More severe hidrosis, no cysts. |
Treatment
No curative treatment exists for SSPS; management is symptomatic and multidisciplinary.
- Keratoderma: Topical keratolytics (urea, salicylic acid), emollients; oral retinoids (acitretin) for severe cases; excision of syringofibroadenomas if symptomatic.
- Hidrocystomas: Surgical excision, electrocautery, or CO2 laser ablation for cosmetic/functional relief; recur often.
- Dental: Prosthodontics, implants for hypodontia.
- Hair/nails: Topical minoxidil or biotin unproven; supportive care.
- Malignancy surveillance: Regular skin exams for skin cancers.
Genetic counseling is recommended for families.
Outcome
SSPS follows a relatively benign course with normal lifespan, though quality of life declines with age due to progressive cysts and keratoderma. Early intervention mitigates complications; long-term follow-up is essential for tumor surveillance and symptom control. Reported cases show survival into adulthood without systemic lethality.
Frequently asked questions
Is Schöpf-Schulz-Passarge syndrome curable?
No, it is genetic and incurable, but symptoms can be managed effectively with symptomatic treatments.
At what age does SSPS typically present?
Skin and nail changes start in early teens; eyelid cysts and diagnosis often in adulthood (mean 64 years).
Can SSPS lead to cancer?
Yes, palmoplantar lesions may develop squamous cell carcinoma; other adnexal tumors reported.
Is genetic testing available for SSPS?
Yes, sequencing of WNT10A confirms diagnosis.
Does SSPS affect sweating or other glands?
Sweating is typically normal, distinguishing it from hypohidrotic ectodermal dysplasias.
References
- Schöpf–Schulz–Passarge syndrome — Wikipedia. 2023. https://en.wikipedia.org/wiki/Sch%C3%B6pf%E2%80%93Schulz%E2%80%93Passarge_syndrome
- Schopf–Schulz–Passarge Syndrome — PMC – NIH. 2018-10-26. https://pmc.ncbi.nlm.nih.gov/articles/PMC6232990/
- Schöpf-Schulz-Passarge syndrome — Orphanet. 2013-11. https://www.orpha.net/en/disease/detail/50944
- Schöpf-Schulz-Passarge syndrome — DermNet. Recent. https://dermnetnz.org/topics/schoepf-schulz-passarge-syndrome
- Schöpf-Schulz-Passarge syndrome — VisualDx. Recent. https://www.visualdx.com/visualdx/diagnosis/schopf-schulz-passarge+syndrome?diagnosisId=56835&moduleId=101
- Case of Schöpf–Schulz–Passarge syndrome — Oxford Academic (Clinical and Experimental Dermatology). 2005. https://academic.oup.com/ced/article-abstract/30/5/528/6626926
Similar Articles






Read full bio of medha deb